
Sulfonated Schiff base copper(ii) complexes as efficient and selective catalysts in alcohol oxidation: syntheses and crystal structures†
 *a
Luísa M. D. R. S.
Martins,
*ab
*a
Luísa M. D. R. S.
Martins,
*ab
Abstract
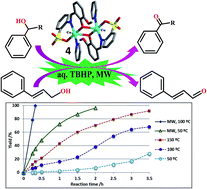
The reaction between 2-aminobenzenesulfonic acid and 2-hydroxy-3-methoxybenzaldehyde produces the acyclic Schiff base 2-[(2-hydroxy-3-methoxyphenyl)methylideneamino]benzenesulfonic acid (H2L·3H2O) (1). In situ reactions of this compound with Cu(II) salts and, eventually, in the presence of pyridine (py) or 2,2′-bipyridine (2,2′-bipy) lead to the formation of the mononuclear complexes [CuL(H2O)2] (2) and [CuL(2,2′-bipy)]·DMF·H2O (3) and the diphenoxo-bridged dicopper compounds [CuL(py)]2 (4) and [CuL(EtOH)]2·2H2O (5). In 2–5 the L2− ligand acts as a tridentate chelating species by means of one of the O-sulfonate atoms, the O-phenoxo and the N-atoms. The remaining coordination sites are then occupied by H2O (in 2), 2,2′-bipyridine (in 3), pyridine (in 4) or EtOH (in 5). Hydrogen bond interactions resulted in R22(14) and in R44(12) graph sets leading to dimeric species (in 2 and 3, respectively), 1D chain associations (in 2 and 5) or a 2D network (1). Complexes 2–5 are applied as selective catalysts for the homogeneous peroxidative (with tert-butylhydroperoxide, TBHP) oxidation of primary and secondary alcohols, under solvent- and additive-free conditions and under low power microwave (MW) irradiation. A quantitative yield of acetophenone was obtained by oxidation of 1-phenylethanol with compound 4 [TOFs up to 7.6 × 103 h−1] after 20 min of MW irradiation, whereas the oxidation of benzyl alcohol to benzaldehyde is less effective (TOF 992 h−1). The selectivity of 4 to oxidize the alcohol relative to the ene function is demonstrated when using cinnamyl alcohol as substrate.
 Please wait while we load your content...
Please wait while we load your content...

