
Synthesis of 2-anilinopyridine–arylpropenone conjugates as tubulin inhibitors and apoptotic inducers†
Abstract
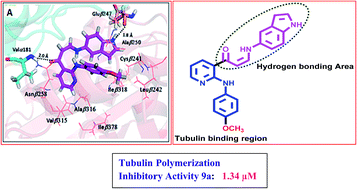
A series of new (Z)-3-(arylamino)-1-(2-(arylamino)pyridin-3-yl)prop-2-en-1-one conjugates 9a–p were synthesized and evaluated for their cytotoxic activity against some human cancer cell lines. Some of the treated compounds like 9a, 9g and 9j showed significant activity with IC50 values ranging from 0.51 to 1.29 μM. Flow cytometry results revealed that for A549 cells these compounds caused accumulation of cells in the G2/M phase. Interestingly, compound 9a demonstrated a considerable inhibitory effect on tubulin polymerization and showed significant inhibition of tubulin polymerization with an IC50 value of 1.34 μM, whereas nocodazole showed antitubulin activity with an IC50 value 2.64 μM. Further, Hoechst staining and activation of caspase-3 suggested that these conjugates induced cell death by apoptosis. Fluorescence based competitive colchicine binding assay and docking studies suggest that these conjugates 9a and 9g bind to the tubulin perfectly at the colchicine binding site.
 Please wait while we load your content...
Please wait while we load your content...

