
Mesoporous carbon/CuS nanocomposites for pH-dependent drug delivery and near-infrared chemo-photothermal therapy
Abstract
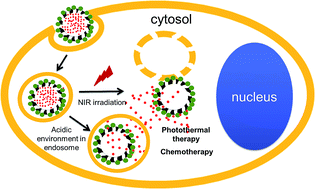
Spurred by the recent development in nanotechnology, multi-functional therapeutic platforms have emerged as promising anti-cancer treatments for their combinational effects. In this paper, we report a novel drug delivery system composed of mesoporous carbon nanospheres (MCN) of 150 to 200 nm in diameter capped with copper sulfide (CuS) nanoparticles (NPs). MCNs can efficiently load doxorubicin (DOX, an anti-cancer drug) due to their hollow and porous structures as well as the π–π stacking interactions between MCN and DOX. DOX is retained in MCN under a basic and physiological environment, but releases rapidly under an acidic environment in its ionized state. Due to the intrinsic near-infrared (NIR) absorption and photothermal conversion ability of copper sulfide nanoparticles, heat is generated for killing tumor cells as well as stimulating DOX release upon NIR irradiation. Thus, this complex (MCN–CuS) exhibits efficient drug loading, low pre-release, temperature-, NIR- and pH-responsive DOX release, and combined antitumor activity.
 Please wait while we load your content...
Please wait while we load your content...

