
Highly active spherical amorphous MoS2: facile synthesis and application in photocatalytic degradation of rose bengal dye and hydrogenation of nitroarenes†
Abstract
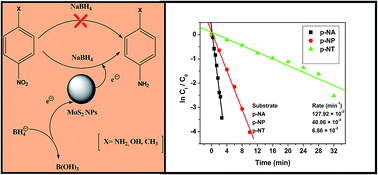
Herein, we developed a facile method to prepare amorphous spherical MoS2 via a simple solvothermal decomposition of a precursor complex MoO2(acda)2 (Hacda = 2-aminocyclopentene-1-dithiocarboxylic acid) in the presence of triethylenetetramine (TETA) as a solvent at 200 °C in an inert atmosphere. The as-obtained product was characterized by X-ray diffraction analysis (XRD), electron diffraction X-ray spectroscopy (EDX), transmission electron microscopy (TEM), and UV-vis spectroscopic techniques. The transmission electron microscopic study ascertains the amorphous particles to be of spherical structure. The amorphous MoS2 has shown photocatalytic activity for the degradation of rose bengal (RB) dye under visible light illumination. The kinetics of the decomposition process was also investigated and found to show the pseudo-first-order reaction kinetics with rate constants of 5.0 × 10−2 min−1. Furthermore, amorphous MoS2 was found to be highly effective in catalyzing the reduction of a series of nitroarenes to the corresponding anilines by an eco-friendly protocol.
 Please wait while we load your content...
Please wait while we load your content...

