
Enantioselective synthesis of mosquito oviposition pheromone and its epimer from a naturally occurring fatty acid†
Abstract
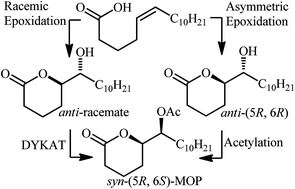
The synthesis of Mosquito Oviposition Pheromones (MOP) (5R,6S)-5-acetoxy-6-hexadecanolide and its unnatural (5R,6R)-diastereomer in 68% and 54% overall yield by a route involving an organocatalyzed epoxidation of naturally occurring cis-5-hexadecenoic acid and diastereodivergent esterification is reported. The investigation of a dynamic kinetic asymmetric transformation (DYKAT) as an alternate strategy for preparing the target MOPs is also discussed, however this approach was unsuccessful due to the formation of a ketone by-product that inhibited the lipase mediated acetylation step of the DYKAT process.
 Please wait while we load your content...
Please wait while we load your content...

