
Protoporphyrin incorporated alginate hydrogel: preparation, characterization and fluorescence imaging in vivo†
Abstract
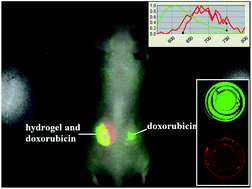
Fluorescence imaging provides a non-invasive tool for locating, monitoring and tracking implanted biomaterials in situ. Since alginate hydrogels have been widely applied as scaffolds for tissue engineering, delivery carriers or extracellular matrices, the in vivo status of alginate hydrogels is necessary and important to their applications. Herein, a protoporphyrin incorporated alginate hydrogel was prepared using PEG as a linker. The structure and properties were investigated with rheological analysis, thermogravimetric analysis, and UV-visible and fluorescence spectrophotometry. Hydrogel erosion and drug delivery in vivo were monitored and tracked using a multispectral fluorescence imaging system with nude mice as models. The results show that the protoporphyrin incorporated alginate hydrogel exhibits fluorescence ability in vivo with good biocompatibility. With the guidance of fluorescence imaging, the in vivo status of hydrogels can be reflected in situ. Protoporphyrin based biomaterials have potential for application in in vivo monitoring and tracking using fluorescence imaging.
 Please wait while we load your content...
Please wait while we load your content...

