
Dissimilatory nitrate reduction to ammonium as an electron sink during cathodic denitrification
Elisa M. Sandera,
Bernardino Virdisab and
Stefano Freguia *ab
*ab
aAdvanced Water Management Centre, The University of Queensland, Level 4, Gehrmann Laboratories Building (60), Brisbane, QLD 4072, Australia. E-mail: s.freguia@awmc.uq.edu.au
bCentre for Microbial Electrochemical Systems, The University of Queensland, Gehrmann Building, Brisbane, Queensland 4072, Australia.
First published on 8th October 2015
Abstract
Dissimilatory nitrate reduction to ammonium (DNRA) is an undesired pathway occurring simultaneously to denitrification in natural environments as well as engineered systems aimed at biological nitrate reduction/removal. Ammonium formation has previously been detected in cathodic compartments of bioelectrochemical systems performing denitrification, although reported concentrations are generally very low. In order to demonstrate and quantify the occurrence of DNRA from nitrate in a mixed culture denitrifying cathodic biofilm, a carbon cloth working electrode was inoculated with a denitrifying microbial community and poised at −0.9 V vs. standard hydrogen electrode, while nitrate (20 mg L−1 NO3−-N) was continuously fed at an HRT of 10 hours. Results showed that more than 40% of nitrogen added as nitrate was converted via DNRA when the biofilm was at initial stages of development. However, ammonium generation decreased to approximately 5% at later stages of development (7 months of operation), indicating that biofilm age plays a key role on biological pathways occurring during cathodic nitrate reduction. A closer insight revealed that the occurrence of DNRA is linked to cathodic coulombic efficiency: at low efficiency, a large fraction of the incoming electrons are converted to hydrogen or other reduced compounds within the biofilm, increasing the driving force for DNRA; at high coulombic efficiency, lower reducing power availability leads to nitrogen gas as preferred reduction product.
Introduction
Nitrogen cycling in natural environments involves a series of different biological reactions, including aerobic oxidation of ammonium during nitrification (i.e., the sequential ammonium and nitrite oxidation to nitrate), followed by the reduction of nitrate to dinitrogen gas (denitrification) in the absence of oxygen.1,2 Another possible but less studied pathway for nitrate reduction is the dissimilatory nitrate reduction to ammonium (DNRA), whereby nitrate is converted directly to ammonium under anoxic conditions.1 It was previously shown that bacteria able to perform DNRA can be isolated from nitrate-contaminated water and soil.3 In addition, this phenomenon is also naturally observed in marine and freshwater sediments,4–6 accounting for up to 30% of nitrate reduction processes, thereby slowing down nitrogen removal through denitrification in such systems.6In engineered systems aimed at wastewater treatment, the occurrence of the DNRA pathway can represent an important challenge towards nitrogen removal. As an undesired pathway, it leads to a high percentage of the nitrogen being retained in the water undergoing denitrification,7 as the regenerated ammonium does not further react in the anoxic treatment stages.
Bioelectrochemical systems (BESs) are recently being studied as a novel technology for denitrification.8,9 These are systems that rely on reactions occurring at inert surfaces of conductive materials (that is, electrodes), in which attached microorganisms perform oxidation/reduction processes as a consequence of the difference in potential between electrode and substrate.10 Denitrification reactions within BES typically occur at cathodic electrode surfaces, via (1) direct transfer of electrons from electrode to microorganisms or (2) electrochemically generated H2 that is utilised as an electron donor for microbial reactions.11–14 Much research has been done for the applicability of cathodic reactions in BESs for the removal of nitrate from waters/wastewaters in the absence of significant amounts of organic matter, such as groundwater,.8,15 Overall, this bioelectrochemical autotrophic denitrification is advantageous over traditional heterotrophic denitrification due to less biomass formation (avoiding clogging of the reactors), and due to the fact that no external organic matter is required in the process, which eliminates the need of supply and on-site storage of chemicals and avoids further carbon contamination into watercourses.16,17 Similarly to what was observed in natural environments, DNRA pathway was shown to account for approximately 34% of cathodic nitrate/nitrite reduction in a fundamental study with pure culture of Pseudomonas alcaliphila.18 Furthermore, ammonium nitrogen was also previously detected in low concentrations in some bioelectrochemical systems (BESs) performing denitrification.13,19–21 However, to our best knowledge, DNRA was never quantitatively characterised in a mixed culture denitrifying cathodic biofilm. Therefore, the aim of this work is to demonstrate and quantify the occurrence of ammonium generation from nitrate in mixed culture denitrifying cathodic biofilm performing autotrophic denitrification.
Material and methods
Bioelectrochemical setup and media composition
A bioelectrochemical cell was constructed with a modified glass bottle (working volume capacity of 0.25 L), in which a glass tube (0.008 L volume capacity) was inserted. The main chamber of the reactor hosted the cathodic (nitrate reducing) electrode and was separated from the smaller anodic compartment by a Cation Exchange Membrane (Ultrex CMI-7000, Membranes International Inc., USA). A Tedlar gas bag with 3 L volume capacity (SKC, USA) was attached to the headspace of the cathodic chamber to avoid overpressure. The anodic chamber was instead exposed to air and hosted the anodic electrode for the oxidation of water.A standard three-electrode setup was used for the experiments and each Working Electrode (WE) consisted of two pieces of plain carbon cloth (Fuel Cell Store, USA) placed in parallel and totalizing 35.2 cm2 projected surface area. Current collection was guaranteed with the use of Ti mesh and a Ti wire. The carbon cloth WE was pre-cleaned with isopropanol (50%) for 4 hours with agitation to remove impurities that could be present on the electrode surface, and then rinsed with reverse osmosis water. Both WE and a reference electrode (Ag/AgCl, +0.197 V vs. SHE) were inserted into the main chamber, while a Counter Electrode (CE) consisting of a Pt wire was inserted in the anodic chamber. The electrodes were connected to a multichannel potentiostat (CHI Instruments, USA) for cathodic potential control.
The cathodic medium consisted of 6 g L−1 Na2HPO4, 3 g L−1 KH2PO4, 0.1 g L−1 MgSO4·7H2O, 0.015 g L−1 CaCl2·2H2O, 1 g L−1 NaHCO3, 20 mg L−1 NO3−-N and trace elements solution as previously described.22 The cathodic medium was previously autoclaved and sparged with N2 for 30 minutes to remove oxygen. The anolyte was composed of 6 g L−1 Na2HPO4 and 3 g L−1 KH2PO4 and operated abiotically at all times.
Reactor inoculation and operation
The reactor was mostly operated under potentiostatic mode, with cathodic medium being continuously mixed with a magnetic stirrer to avoid diffusion limitations, except when performing cyclic voltammetry (CV) experiments. A fixed potential of −0.9 V vs. standard hydrogen electrode (SHE) was applied to the cathode electrode unless stated otherwise. A denitrifying microbial community previously grown heterotrophically in a fed-batch reactor was used as inoculum source for the cathodic denitrification experiments. After inoculating the cathode with a small amount of 13 mg as COD of the specified denitrifying culture, the BES reactor was operated in batch mode with no media replacement for 22 days (adaptation period). Exceptionally during this adaptation period (in order to gradually adapt the microbes to autotrophic conditions), periodic additions of nitrate (20 mg L−1 NO3−-N) as electron acceptor, simultaneously of acetate and methanol as carbon sources, were carried out whenever concentrations of nitrate were detected to be lower than 5 mg L−1 NO3−-N, corresponding to days 0 (simultaneously to inoculation), 2, 3, 5, 6, 8 and 10. Carbon sources were added at decreasing concentrations, corresponding to COD/N ratios of approximately 8.2, 5.4, 2.9, 1.8, 1.1, 0.5 and lastly again 0.5, respectively, until day 10 as exposed above. Therefore, organic matter was no longer added to the reactor after day 10. After the adaptation period, the reactor was further operated in sequential batch mode with initial 20 mg L−1 NO3−-N and 50% of the media being replaced weekly, in order to keep lower ammonium concentrations in the reactor and avoid washing out the biomass (enrichment period, days 23–89). Finally, in order to guarantee a constant supply of nitrate and removal of by-products while still enabling enrichment of electroactive microbial consortia in the biofilm, the reactor was switched to a continuous feed mode (approximately 0.6 L d−1 flow rate, corresponding to 10 hours hydraulic retention time) from day 90 onwards, with the same nitrate concentration as previously specified. Cyclic voltammetry (CV) experiments were carried out at different phases of reactor operation in the presence of nitrate, which guaranteed turnover conditions without limiting electron acceptors. CVs were performed without agitation, at a scan rate of 1 mV s−1. The applied potential range was from −1.3 to −0.3 and −1.1 to −0.3 V vs. SHE for blank (control) and biofilm operation CVs, respectively.Reactor operation during batch tests
A preliminary assessment of the biofilm activity was done at approximately 1 month of operation, during the enrichment phase as specified previously. After the reactor reached a steady current under continuous feed mode, specific batch tests with initial concentration of roughly 20 mg L−1 NO3N were then carried out for the assessment of DNRA at 5 and 7 months of operation, to evaluate whether the development stage of the biofilm plays a role on ammonium formation. Prior to each individual test, the feed was interrupted and medium was replaced completely with fresh medium while sparging the reactor with N2. The reactor was otherwise kept in continuous feed mode between the tests.Analytical methods and calculations
Liquid samples collected from the catholyte were filtered with a 0.22 μm filter (Millipore Express, USA). The concentrations of NO3−-N, NO2−-N and NH4+-N were determined using a Lachat QuikChem8000 Flow Injection Analyzer (Lachat Instruments, Milwaukee, USA).Coulombic efficiency (CE) of batch tests was calculated with the following formula:

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C per mol e−). Although nitric oxide (NO) and nitrous oxide (N2O) are also intermediate compounds during denitrification, they were not included in the above formula. Firstly, nitric oxide tends to be consumed upon its formation and the accumulation of this compound can therefore be considered negligible as previously demonstrated.2,8,23 Secondly, although approximately 9% of losses were reported to be due to accumulation of N2O in batches carried out at −0.2 V vs. SHE, the percentage of electrons transferred as current and lost as N2O was also shown to considerably decrease with decreasing cathodic potentials.23 Thus, since the applied potential within this study (−0.9 V vs. SHE) is considerably lower than those reported in the referred study, N2O accumulation can also be considered negligible.
485 C per mol e−). Although nitric oxide (NO) and nitrous oxide (N2O) are also intermediate compounds during denitrification, they were not included in the above formula. Firstly, nitric oxide tends to be consumed upon its formation and the accumulation of this compound can therefore be considered negligible as previously demonstrated.2,8,23 Secondly, although approximately 9% of losses were reported to be due to accumulation of N2O in batches carried out at −0.2 V vs. SHE, the percentage of electrons transferred as current and lost as N2O was also shown to considerably decrease with decreasing cathodic potentials.23 Thus, since the applied potential within this study (−0.9 V vs. SHE) is considerably lower than those reported in the referred study, N2O accumulation can also be considered negligible.
Results and discussion
DNRA during cathodic biofilm adaptation
The behaviour of the reactor during the start-up (adaptation) period is presented in Fig. 1. The current started to increase upon inoculation with denitrifying microbial community (Fig. 1A). The nitrate pulse (20 mg L−1 N) was completely consumed after the first two days of operation and was periodically added in the reactor as indicated in Fig. 1B. Simultaneously to the additions of nitrate, acetic acid and methanol were added at decreasing concentrations as previously detailed in the Materials and methods section. Both organic compounds were no longer present in the reactor after 17 days. Nitrite was sporadically detected in the reactor during the adaptation period at maximum concentration of approximately 2.4 mg L−1 N, however this compound was consumed afterwards and there was no nitrite left in the reactor at the end of the adaptation period (Fig. 1C). The figure also shows that ammonium starts forming approximately after two days of operation and accumulates to a concentration of 27 mg L−1 N in 22 days. The formation of ammonium during this stage could primarily be linked to the presence of acetate and methanol in relatively high concentrations during initial stages of the adaptation phase. As it has been previously shown, high C/N ratios (which can be translated as the ratio of electron donors to acceptors) due to high organic matter concentrations favours the DNRA pathway during heterotrophic nitrate reduction.24 However, the observation that ammonium formation/accumulation still occurs in the absence of organic electron donors after day 17 indicates that DNRA is possible also in autotrophic systems with electrons provided from the cathode. | ||
| Fig. 1 Inoculation/adaptation period (22 days). (A) Current profile; (B) NO3−-N profile and (C) NO2−-N and NH4+-N concentrations, respectively. Single black arrow at day zero indicate inoculation time whereas red arrows between days 0 and 10 indicate additions of nitrate simultaneously to decreasing concentrations of organic matter (acetic acid and methanol). Asterisks indicate additions of nitrate only. | ||
The DNRA pathway is generally not considered in most cathodic denitrification studies due to the absence or very low concentrations of ammonium usually detected within the denitrifying cathodes.23,25 However, our results indicate that it is in fact happening in mixed culture denitrifying cathodes, which corroborates a previous publication that reported detection of ammonium in denitrifying microbial fuel cell (MFC) inoculated with non-adapted microbial community.13 Therefore, as nitrate is partially converted to ammonium, the corresponding amount of nitrogen cannot be removed from the solution under the anoxic conditions encountered in this system.
It has previously been reviewed that DNRA can be favoured over denitrification when more reducing environmental conditions occur.26 A study done with pure culture of Pseudomonas alcaliphila showed that proportionally more electrons are transferred from nitrate to ammonium when applying a lower potential such as −0.9 V, as opposed to +0.1 or −0.1 V vs. SHE.27 Although the effects of cathodic potential on ammonium formation were not evaluated herein, those previously reported results give an insight on the reasons why some previously studied BES operating in MFC mode (with much higher cathodic potential) did not detect any ammonium formation,23 whereas up to 4.1 mg L−1 ammonium accumulated at the end of preliminary batches in the present study (data not shown).
DNRA is linked to biofilm development stage and electron donor availability
Cyclic voltammetry performed before inoculation of the reactor show a lack of significant reductive current at −0.9 V vs. SHE, indicating absence or very low catalytic formation of hydrogen under abiotic conditions at this potential (Fig. 2). The onset of cathodic nitrate reduction shifted towards more positive potential values as the denitrification activity developed, as shown in Fig. 2.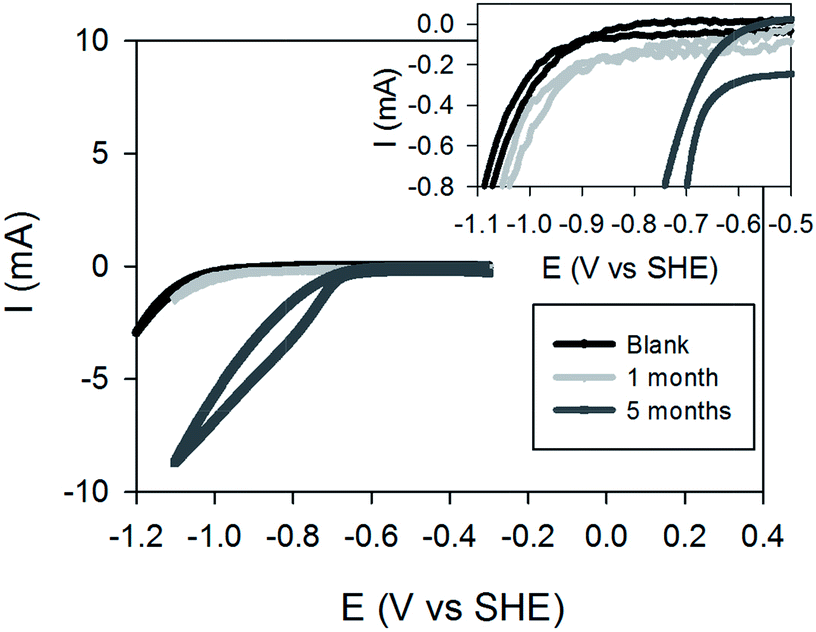 | ||
| Fig. 2 Cyclic voltammograms performed in turnover conditions at different times of cathodic biofilm development (0, 1 and 5 months). Scan rate 1 mV s−1. The inset indicates onsets of catalytic currents. | ||
Current and nitrogen species time profiles during the batch tests performed at different operational stages are shown in Fig. 3. The current increased over time when batches were carried out in the young biofilm (Fig. 3B). However, the current did not seem to be affected by decreasing nitrate concentrations. Furthermore, the current tended to be more stable and higher in magnitude for the batches carried out with the 5 and 7 months old biofilm (Fig. 3D and F). Noteworthy, the applied cathodic potential of −0.9 V vs. SHE is lower than the theoretical hydrogen evolution potential (−0.41 V vs. SHE), hence hydrogen production is expected. A few studies have previously shown hydrogen formation at cathodic surfaces in bioelectrochemical systems,28,29 and its production was also shown to be enhanced over time with the use of microorganisms as catalysts when applying the potential of −0.75 V vs. SHE.30 In addition, electrons delivered through a biocatalized cathode were also previously shown to be completely recovered as hydrogen (100% cathodic hydrogen efficiency) if a negligible diffusion is occurring through the membrane.29 Thus, although hydrogen measurements of liquid and gas phases were not done in this work, the current behaviour presented herein corroborates an increasing H2 formation over time as reported in the above cited literature, which is further supported by the CVs shown in Fig. 2.30
 | ||
| Fig. 3 Nitrogen species and current profile during batch tests at −0.9 V vs. SHE carried out with initial concentration of 20 mg L−1 NO3−-N at different biofilm development stages. (A and B) 1 month old biofilm tests (n = 2); (C and D) 5 month old biofilm (n = 2); (E and F) 7 month old biofilm (n = 3). Nitrogen concentrations are plotted as averages (±standard deviations), whereas current profiles are plotted for each individual test (T1, T2 and/or T3, depending whether they were done in duplicates or triplicates). | ||
Reduction of nitrate from 20 mg L−1 NO3−-N to approximately 8 mg L−1 required 2 days of operation when the biofilm was 1 month old, whereas approximately the same amount of nitrogen was reduced within 12 hours during the batch tests carried out at 5 and 7 months of biofilm operation. Nitrite was detected in low concentrations during the 1 month tests and was found to be below detection limits at the end of those batches and also at all times during the batches carried out in months 5 and 7. Furthermore, the amount of ammonium formed during batches at different biofilm ages decreased from 9.6 ± 3.5 to 3.80 ± 0.3 and 0.76 ± 0.4 mg L−1, respectively at 1, 5 and 7 months of operation.
A more detailed analysis of batches carried out at different times of operation indicates a metabolic shift occurring within the biofilm, as shown in Fig. 4. As it can be noticed in the bar chart, the percentage of nitrate converted to ammonium tended to decrease over time of biofilm operation/adaptation. An average of 47.8% ± 19.7% of all reduced NO3−-N was converted to ammonium within the young biofilm, whereas only 5.8% ± 2.8% was converted via DNRA pathway within the 7 month old biofilm. This fact demonstrates that a long time period is required for the denitrifying cathodic biofilm development. A 46 days start-up time was previously reported for a cathodic biofilm31 and a long maturation time of months instead of weeks was also observed in a study evaluating differences of electrochemical impedance over time for anodic biofilms inoculated with non-adapted biomass.32
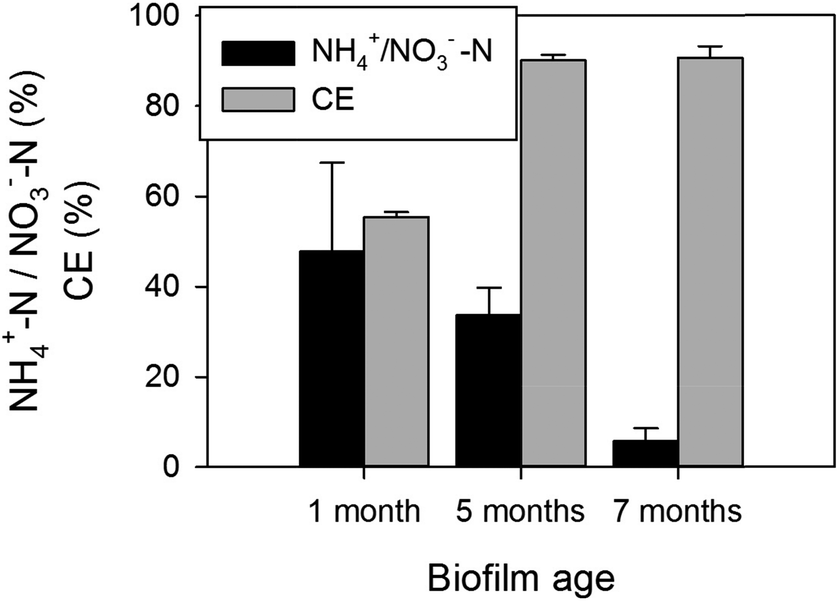 | ||
| Fig. 4 Calculated coulombic efficiency (CE) and ratio of NH4+-N formed to the total amount of nitrate nitrogen consumed at the end of batches carried out with the biofilm aged at 1 (n = 2), 5 (n = 2) and 7 months old (n = 3). | ||
The previously reported fact that more reductive environmental conditions play a role on the occurrence of DNRA helps understanding the higher than usual ammonium formation in this study. However, as the potential applied herein was constant over the whole study period, this hypothesis does not explain the decreasing ammonium formation observed over time. It was previously shown that a high C/N ratio (translated to electron donor/acceptor ratio in autotrophic conditions) stimulated the enrichment of DNRA bacteria, that were able to convert up to 90% of all nitrate into ammonium in a chemostat.24 In addition, the electron donor/acceptor ratio was in fact found to play a role alongside environmental conditions in DNRA regulation.26 Furthermore, the effect of electron donor-to-acceptor ratio was also confirmed in BES pure culture studies.18,27 An assessment of coulombic efficiencies (CE) in the present study indicated that lower efficiencies of approximately 55.4% ± 1.1% were obtained at an early stage of operation, as opposed to coulombic efficiencies higher than 90% at later stages of biofilm development (see Fig. 4). A low coulombic efficiency implies an excess of electrons being transferred from the cathode relatively to those used in the reduction of nitrate, and therefore an excess of free hydrogen was likely to have occurred at early stages. When assuming that (1) the excess electrons delivered from the cathode must necessarily go into hydrogen generation30,33 as explained previously, and (2) 1 mmol H2 requires 2 mmol electrons transferred from the cathode, then an averaged hydrogen production can be calculated from the charge transferred during the batches to be 0.021 (±0.001) and 0.055 (±0.002) mmol H2 per hour for 1 and 7 months operation respectively, which shows that hydrogen formation was actually considerably smaller in the first month compared to latter stages. However, an important factor for consideration is the ability of the biofilm to consume electrons for denitrification. Since the biofilm was still not fully developed at 1 month operation, its ability to carry out autotrophic denitrification and consume that hydrogen was very poor as indicated by the low nitrate reduction rate (5.1 ± 0.3 g N m−3 NCC d−1 ± SD). Therefore, it resulted in free H2 within the biofilm which translated into low CE. On the other hand, although calculated H2 production was higher at latter stages, as explained above, nitrate reduction rate was considerably faster (26.0 ± 1.4 g N m−3 NCC d−1 ± SD). Therefore, this relation between electrons delivered as H2 and consumed during nitrate reduction was reflected in the higher CE obtained at later stages of operation, and suggesting that the shortage of electrons available as free hydrogen led to a restriction in the DNRA pathway in the 7th month operation. The high electron donor (hydrogen)-to-nitrate ratio established at low CE would have led to DNRA as preferential pathway. Noteworthy, the complete denitrification pathway from nitrate to nitrogen gas requires only 5 electrons for the reduction of each nitrate molecule, whereas the DNRA pathway requires a total of 8 electrons. Therefore, it is understandable that a bigger availability of electrons in the system would enable the formation of ammonium in detriment of the denitrification pathway. An important factor for consideration is the possibility of a shift in microbial community composition over time. An analysis of cathodic biofilm community structure over time is warranted for future work, in order to identify the main drivers and the dynamics of the DNRA process in BES.
Conclusions
Dissimilatory nitrate reduction to ammonium was shown to be a possible electron sink during denitrification in mixed culture cathodic denitrifying biofilms operated at −0.9 V vs. SHE. This study also demonstrated that ammonium generation via DNRA pathway is dependent on biofilm age and can decrease over time of operation, likely due to reduced hydrogen availability within the biofilm. The results presented here stress the importance of having a fully developed biofilm for cathodic reduction of nitrate from contaminated water/wastewaters, in order to avoid unwanted production of ammonium and the consequent retainment and/or discharge of nitrogen compounds.Acknowledgements
This work was funded by the Australian Research Council, grant ARC-DP120104415. BV acknowledges the financial support for CEMES through The University of Queensland.References
- W. G. Zumft, Microbiol. Mol. Biol. Rev., 1997, 61, 533–616 CAS.
- D. Richardson, H. Felgate, N. Watmough, A. Thomson and E. Baggs, Trends Biotechnol., 2009, 27, 388–397 CrossRef CAS PubMed.
- R. Seenivasagan, S. Rajakumar, R. Kasimani and P. M. Ayyasamy, Prep. Biochem. Biotechnol., 2014, 44, 586–597 CrossRef CAS PubMed.
- J. T. Scott, M. McCarthy, W. Gardner and R. Doyle, Biogeochemistry, 2008, 87, 99–111 CrossRef CAS.
- F. Sgouridis, C. M. Heppell, G. Wharton, K. Lansdown and M. Trimmer, Water Res., 2011, 45, 4909–4922 CrossRef CAS PubMed.
- G. D. Song, S. M. Liu, H. Marchant, M. M. M. Kuypers and G. Lavik, Biogeosciences, 2013, 10, 6851–6864 CAS.
- B. Kløve, A. K. Søvik and L. Holtan-Hartwig, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2005, 40, 1381–1401 CrossRef PubMed.
- S. Puig, M. Coma, J. Desloover, N. Boon, J. Colprim and M. D. Balaguer, Environ. Sci. Technol., 2012, 46, 2309–2315 CrossRef CAS PubMed.
- B. Virdis, K. Rabaey, R. A. Rozendal, Z. Yuan, Y. Mu and J. Keller, J. Biotechnol., 2010, 150, 153–154 CrossRef PubMed.
- K. Rabaey, J. Rodriguez, L. L. Blackall, J. Keller, P. Gross, D. Batstone, W. Verstraete and K. H. Nealson, ISME J., 2007, 1, 9–18 CrossRef CAS PubMed.
- B. Virdis, K. Rabaey, Z. Yuan and J. Keller, Water Res., 2008, 42, 3013–3024 CrossRef CAS PubMed.
- T. Watanabe, H. Motoyama and M. Kuroda, Water Res., 2001, 35, 4102–4110 CrossRef CAS.
- P. Clauwaert, K. Rabaey, P. Aelterman, L. De Schamphelaire, T. H. Pham, P. Boeckx, N. Boon and W. Verstraete, Environ. Sci. Technol., 2007, 41, 3354–3360 CrossRef CAS.
- Y. Zhao, B. Zhang, C. Feng, F. Huang, P. Zhang, Z. Zhang, Y. Yang and N. Sugiura, Bioresour. Technol., 2012, 107, 159–165 CrossRef CAS PubMed.
- N. Pous, S. Puig, M. Coma, M. D. Balaguer and J. Colprim, J. Chem. Technol. Biotechnol., 2013, 88, 1690–1696 CrossRef CAS PubMed.
- S. J. Ergas and A. F. Reuss, J. Water Supply: Res. Technol.--AQUA, 2001, 50, 161–171 CAS.
- J. van Rijn, Y. Tal and H. J. Schreier, Aquacultural Engineering, 2006, 34, 364–376 CrossRef PubMed.
- W. Su, L. Zhang, D. Li, G. Zhan, J. Qian and Y. Tao, Biotechnol. Bioeng., 2012, 109, 2904–2910 CrossRef CAS PubMed.
- S. Kondaveeti and B. Min, Bioprocess Biosyst. Eng., 2013, 36, 231–238 CrossRef CAS PubMed.
- B. Huang, H. Feng, M. Wang, N. Li, Y. Cong and D. Shen, Bioresour. Technol., 2013, 132, 91–98 CrossRef CAS PubMed.
- C. Fang, B. Min and I. Angelidaki, Appl. Biochem. Biotechnol., 2011, 164, 464–474 CrossRef CAS PubMed.
- H. Lu, A. Oehmen, B. Virdis, J. Keller and Z. Yuan, Water Res., 2006, 40, 3838–3848 CrossRef CAS PubMed.
- B. Virdis, K. Rabaey, Z. Yuan, R. A. Rozendal and J. Keller, Environ. Sci. Technol., 2009, 43, 5144–5149 CrossRef CAS.
- E. M. van den Berg, U. van Dongen, B. Abbas and M. C. van Loosdrecht, ISME J., 2015, 9, 2153–2161 CrossRef CAS PubMed.
- N. Pous, S. Puig, M. Dolors Balaguer and J. Colprim, Chem. Eng. J., 2015, 263, 151–159 CrossRef CAS PubMed.
- T. Rütting, P. Boeckx, C. Müller and L. Klemedtsson, Biogeosciences, 2011, 8, 1779–1791 Search PubMed.
- W. Zhang, Y. Zhang, W. Su, Y. Jiang, M. Su, P. Gao and D. Li, J. Environ. Sci., 2014, 26, 885–891 CrossRef CAS.
- R. A. Rozendal, A. W. Jeremiasse, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2007, 42, 629–634 CrossRef.
- R. A. Rozendal, H. V. M. Hamelers, R. J. Molenkamp and C. J. N. Buisman, Water Res., 2007, 41, 1984–1994 CrossRef CAS PubMed.
- L. Jourdin, S. Freguia, B. C. Donose and J. Keller, Bioelectrochemistry, 2015, 102, 56–63 CrossRef CAS PubMed.
- J. Desloover, S. Puig, B. Virdis, P. Clauwaert, P. Boeckx, W. Verstraete and N. Boon, Environ. Sci. Technol., 2011, 45, 10557–10566 CrossRef CAS PubMed.
- A. P. Borole, D. Aaron, C. Y. Hamilton and C. Tsouris, Environ. Sci. Technol., 2010, 44, 2740–2745 CrossRef CAS PubMed.
- Y. Sakakibara and M. Kuroda, Biotechnol. Bioeng., 1993, 42, 535–537 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |