
A new fabrication of AgX (X = Br, I)–TiO2 nanoparticles immobilized on polyacrylonitrile (PAN) nanofibers with high photocatalytic activity and renewable property
Dandan
Yu
,
Jie
Bai
*,
Haiou
Liang
,
Junzhong
Wang
and
Chunping
Li
Chemical Engineering College, Inner Mongolia University of Technology, Huhhot 010051, People's Republic of China. E-mail: baijie@imut.edu.cn; Fax: +86 471 6575722; Tel: +86 471 6575722
First published on 21st October 2015
Abstract
A highly efficient visible light-driven AgX–TiO2/PAN (X = Br, I) photocatalyst was synthesized by means of a combination of the electrospinning technique, solvothermal synthesis, physical adsorption and gas/solid reaction. The components, morphological and optical properties of the photocatalysts were characterized by X-ray diffraction (XRD), field-emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), UV-vis diffuse reflectance spectroscopy (DRS) and Fourier transform infrared spectroscopy (FTIR). The as-prepared composites exhibited excellent photocatalytic efficiency for the degradation of methyl orange (MO), methylene blue (MB), acid red 18, sodium fluorescein, xylenol orange and phenol under visible light irradiation. Compared with pure PAN, AgX/PAN and TiO2/PAN, AgX–TiO2/PAN showed much higher photocatalytic activity in degrading MO. In addition, AgX–TiO2/PAN had a certain photochemical stability and could be regenerated easily. The application of PAN nanofibers made it easy to separate the catalysts from an aqueous solution without any loss. The degradation of MO in the presence of different scavengers suggested that holes and ˙O2− were the main reactive species and holes played the predominant role. Thus, a possible two-stage photocatalytic mechanism associated with AgX–TiO2/PAN was proposed.
Introduction
Currently, semiconductor photocatalysis has attracted remarkable attention in solving environmental and energy problems.1–4 From the semiconductor photocatalysts investigated, TiO2 is considered to be a desirable material for the elimination of organic pollutants due to its high activity, stability, low cost and nontoxicity.5–7 However, the practical application of TiO2 has been greatly hampered by the inefficient utilization of solar energy because of its wide band gap (anatase, 3.2 eV; rutile, 3.0 eV) and the rapid recombination of photogenerated electron–hole pairs.5–10 There are some different approaches to be explored to extend the absorption edge of TiO2 into the visible region, such as nonmetal and metal doping,7,8 anionic or cationic doping,10 noble metal deposition11 and coupling with other narrow band gap semiconductors.12Silver halides (AgX, X = Br, I), the important narrow band gap semiconductors, have been recognized as photosensitive materials and extensively used in photographic films.13–15 Under light irradiation, silver halides absorb photons and electron–hole pairs can be liberated.15,16 The photogenerated electrons are easily captured by the Ag+, leading to formation of silver atoms (Ag0).14–16 In general, silver halides are unstable under light irradiation, which greatly affects their applications in photocatalytic reactions. Many researchers have reported that AgX could maintain its stability by coupling with other support materials such as TiO2,16–22 ZnO23–25 and BiOX.5,26,27 Hu et al. have prepared AgI/TiO2 photocatalysts by deposition–precipitation method and the catalyst's activity was maintained effectively for degradation of K-2G after six cycling runs under visible light irradiation without the destruction from AgI.16 According to previous researches, the photosensitivity of AgX makes it feasible synthesize a novel photocatalyst by coupling AgX nanoparticles with TiO2.
In addition, TiO2 particles are easy to agglomerate and difficult to recover after being used for photocatalytic reactions.28,29 To overcome these hurdles, many efforts have been made to immobilize TiO2 nanoparticles on various supporters such as zeolite,31 carbon nanofibers,32 carbon nanotubes33 and graphene.30,34 Owing to the advantages of fine stability and easily availability,36–39 polyacrylonitrile (PAN) nanofibers obtained from an effective and economical electrospinning technique may be promising materials for the immobilization of catalysts as follows: (1) the large specific surface area is beneficial to the high exposure level of photocatalysts nanoparticles and further enhance the photocatalytic activity,28,35,39 (2) the randomly arrayed nanofibers favour the separation, recovery and reuse of photocatalysts,39 (3) PAN nanofibers are hydrophobic with a low density and easily fixed at the proper position of the reactors, which could maximize the efficiency of light utilization by avoiding the hindrance of light penetration.29 Su et al. have successfully fabricated TiO2/PAN hybrid nanofibers by electrospinning and hydrothermal processes.35 The as-prepared catalysts showed the high removal efficiencies of SO2 and NO in the UV light photocatalysis oxidation of flue gas. Therefore, it is a great ideal to prepare a recyclable photocatalyst by immobilizing TiO2 on the surface of PAN nanofibers.
Based on the above statements, a novel photocatalyst AgX (X = Br, I)–TiO2 nanoparticles immobilized on PAN nanofibers were successful prepared through the electrospinning, solvothermal synthesis,29 physical adsorption process and gas/solid reaction.38 In the present work, a series of photocatalysts with different molar ratios between AgX and TiO2 had been fabricated and the photocatalytic activities of the as-prepared catalysts were evaluated by decomposing different organics (methyl orange, acid red 18, methylene blue, xylenol orange, sodium fluorescein and phenol) under visible light irradiation. Moreover, a recycling test was conducted to investigate the photochemical stability and reusability of catalysts and a possible photocatalytic mechanism of the highly enhanced performance was also proposed.
Experimental section
Materials
Polyacrylonitrile (PAN, Mw = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Kunshan Hongyu Plastics Co., Ltd. N,N-Dimethylformamide (C3H7NO, AR, 99.5%) was purchased from Tianjin Fuyu Fine Chemical Co., Ltd. Tetra-n-butyl titanate (Ti(OC4H9)4, AR, 98.5%) was purchased from Xiya Reagent. Acetic acid (CH3COOH, AR, 99.5%) was provided by Beijing Chemicals Co. Silver nitrate (AgNO3, AR, 99.8%), absolute ethyl alcohol (C2H6O, AR, 99.7%) and bromine (Br2, AR, 99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Iodine (I2, AR, 99.8%) was supplied by Tianjin Chemical Reagent Factory. The above chemical reagents were used as received without future treatment.
000) was purchased from Kunshan Hongyu Plastics Co., Ltd. N,N-Dimethylformamide (C3H7NO, AR, 99.5%) was purchased from Tianjin Fuyu Fine Chemical Co., Ltd. Tetra-n-butyl titanate (Ti(OC4H9)4, AR, 98.5%) was purchased from Xiya Reagent. Acetic acid (CH3COOH, AR, 99.5%) was provided by Beijing Chemicals Co. Silver nitrate (AgNO3, AR, 99.8%), absolute ethyl alcohol (C2H6O, AR, 99.7%) and bromine (Br2, AR, 99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Iodine (I2, AR, 99.8%) was supplied by Tianjin Chemical Reagent Factory. The above chemical reagents were used as received without future treatment.
Synthesis of TiO2/PAN hybrid nanofibers
Polyacrylonitrile (PAN) was dissolved in N,N-dimethylformamide (DMF) with vigorous stirring to form a homogeneous 8 wt% PAN/DMF solution for the subsequent electrospinning process. The solution was placed in a glass dropper. A self-made copper ring was wrapped around the top of glass dropper and connected to the anode of a high voltage generator. The applied direct current voltage was 17 kV. A piece of aluminium foil connected to the cathode was placed in 15 cm distance from the tip of the dropper to collect PAN nanofibers. Secondly, 1 ml tetra-n-butyl titanate (Ti(OC4H9)4) and 0.15 ml acetic acid (CH3COOH) were respectively added into 20 ml continuous stirred absolute ethyl alcohol. Then 0.15 g of PAN nanofibers was dispersed into the above mixture solution followed by 24 h physical adsorption. Subsequently, the mixtures were transferred into a 100 ml Teflon-lined stainless autoclave and kept at 180 °C for 9 h. After cooling to room temperature naturally, the products were collected, washed several times with water and dried at 80 °C for 2 h in a vacuum oven. Thus, TiO2/PAN hybrid nanofibers were fabricated.Preparation of AgX–TiO2/PAN (X = Br, I) nanocomposites
AgX–TiO2/PAN (X = Br, I) was synthesized as follows: 0.0250 g AgNO3 was dissolved in 100 ml deionized water under dark condition. Then 0.4 g of TiO2/PAN hybrid nanofibers was added subsequently by physical adsorption for 6 h with stirring constantly. The composites contained Ag+ were filtered, washed three times using deionized water and transferred to a vacuum oven to dry at 90 °C for 3 h. To prepare AgX–TiO2/PAN, the obtained Ag(I)–TiO2/PAN composites were exposed in I2 atmosphere for 48 h (or Br2 atmosphere for 24 h). Finally, they were placed in a vacuum oven to evaporate residual I2 (or Br2) at 90 °C for 4 h. And the as-prepared sample was named AgX (10%)–TiO2/PAN. By changing the dosage of AgNO3 to 0.0050 g, 0.0100 g, 0.0150 g and 0.0200 g, respectively, a series of AgX–TiO2/PAN photocatalysts was prepared and labeled as AgX (y%)–TiO2/PAN, where y% represents the molar ratio of AgX/TiO2. For comparison, AgX/PAN (X = Br, I) was synthesized through similar processes by using 0.0250 g AgNO3 and 0.3 g PAN without solvothermal treatment.Characterization
To study the crystal phase and crystalline of the samples, X-ray diffraction (XRD, Rigaku Ultima IV, Japan) patterns were performed in a range of 2θ from 10° to 90° with a scanning rate of 2° min−1. The morphologies of the products were observed by field-emission scanning electron microscopy (FE-SEM, FEGQUANTAN 650) and transmission electron microscopy (TEM, F20 S-TWIN, Tecnai). The UV-vis diffuse reflectance spectra (DRS) were measured with a Shimadzu UV (3600)-vis spectrophotometer and BaSO4 was used as a reference material. Fourier transform infrared spectra (FTIR) were recorded from KBr pellet in a Nicolet Nexus 670 spectrophotometer with a range of 400–4000 cm−1.Photocatalytic performance
The photocatalytic activities of the as-prepared samples were measured by degrading methyl orange under visible light (λ ≥ 400 nm) irradiation at ambient condition. The light source was a 300 W Xe arc lamp (Beijing Perfectlight Technology Co., Ltd) with a 400 nm cutoff filter. In a typical procedure, 0.2 g photocatalysts were dispersed in 100 ml of a 5 mg l−1 aqueous solution of MO placed in a beaker. Prior to irradiation, the suspension was magnetically stirred in dark for 30 min to establish adsorption–desorption equilibrium. The experiments were carried out under room air-equilibrated conditions. The light was focused onto the breaker. After visible light irradiation at a given time intervals, 3 ml suspensions were sampled and filtered by membrane filters (0.22 μm pore size). The concentrations of dye aqueous solution were analyzed by UV-vis spectrophotometer (UV-1800, Mapada) at a maximum adsorption wavelength of MO (λ = 464 nm). The photocatalytic performance of TiO2/PAN, AgBr/PAN and AgI/PAN was also measured under the same conditions.In order to demonstrate the as-prepared samples could degrade different organic pollutants, the photocatalytic activity of AgX–TiO2/PAN was measured by monitoring the decomposition of five organics (acid red 18, methylene blue, sodium fluorescein, xylenol orange and phenol) aqueous solutions at the same conditions. For detecting the reactive species during photocatalytic degradation, holes (h+), hydroxyl radicals (˙OH), superoxide radical (˙O2−) and 1O2 were investigated by adding 0.2 mM ethylenediamine tetraacetic acid disodium salt (Na2EDTA), 2 mM isopropanol (IPA), 0.2 mM p-benzoquinone (BQ) and 0.2 mM sodium azide (NaN3), respectively. The photocatalytic conditions were similar to the above experiments.
To investigate the photochemical stability of the catalysts, the recycling tests of AgX–TiO2/PAN with the optimum component, which could completely degrade MO, were conducted for five times. At the end of each cycle, the catalysts were collected by filtration, washed with deionized water and dried at 80 °C for 1 h. Then fresh MO solution was mixed with the used catalysts for the next cycle.
Results and discussion
Synthetic procedures
The overall fabrication procedures of AgX–TiO2/PAN (X = Br, I) nanocomposites are illustrated in Scheme 1. It started with the preparation of PAN nanofibers by electrospinning 8 wt% PAN/DMF homogeneous solution, proceeding with dispersing electrospun PAN nanofibers in a Ti(OC4H9)4/ethanol solution and then following to be physical adsorption for 24 h. Then, TiO2 nanoparticles embedded on PAN nanofibers were fabricated by the alcoholysis of Ti(OC4H9)4 during solvothermal process, which promoted the formation of TiO2 with highly crystallization and uniform size. The as-prepared TiO2/PAN hybrid nanofibers were added in an AgNO3 aqueous solution and Ag(I) could be absorbed on their surface. Finally, AgX–TiO2/PAN nanocomposites were fabricated by transforming Ag(I) into AgX through evaporated iodine or bromine.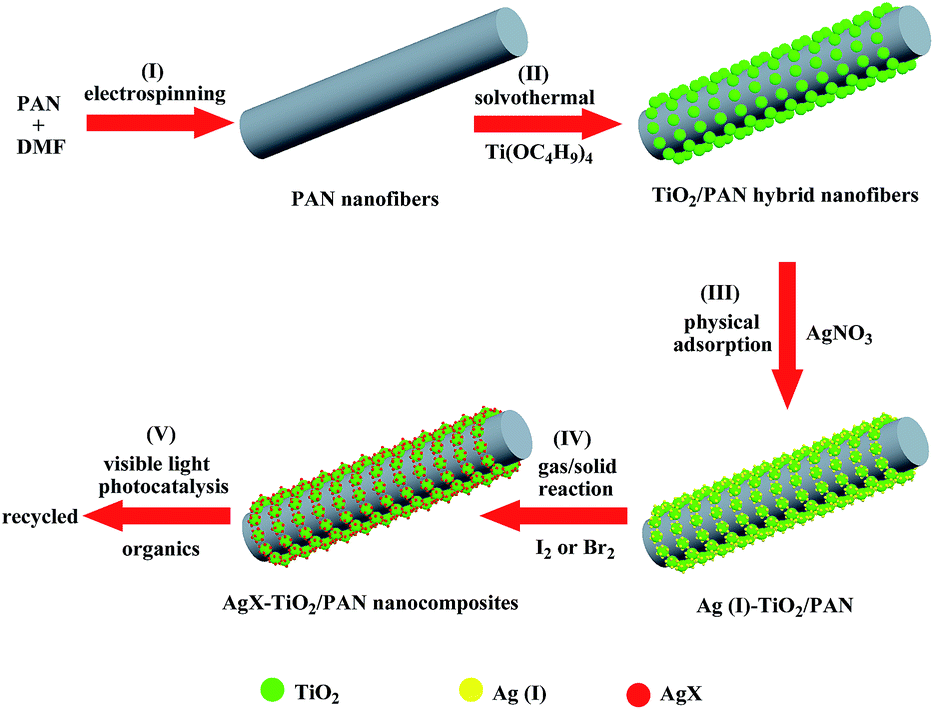 | ||
| Scheme 1 Schematic illustration for the preparation of AgX–TiO2/PAN (X = Br, I) nanocomposites. | ||
Catalysts characterization
The crystalline structure of the as-prepared samples was determined by X-ray diffraction (XRD) measurements. XRD patterns of TiO2/PAN, AgBr (10%)–TiO2/PAN and AgI (10%)–TiO2/PAN are shown in Fig. 1. All samples exhibited the diffraction peaks of anatase TiO2 (JCPDS file no. 21-1272), which proved that the solvothermal treatment was beneficial to the formation of anatase TiO2.29,31,35 The wide peaks appeared in the range of 15° and 20° had been detected, which should be attributed to PAN polymer phase.35 In addition to the diffraction peaks of TiO2 and PAN, the peaks at 2θ = 31.28° and 44.63° were indexed to AgBr (200) and (220) (JCPDS file no. 06-0438),40 while the peak around 2θ = 22.42° should be assigned to β-AgI (100) (JCPDS file no. 09-0374).6,16,20,21 No other diffraction peaks were displayed in the as-prepared AgX (10%)–TiO2/PAN. These results revealed that AgX (X = Br, I) nanoparticles could be synthesized by gas/solid reaction.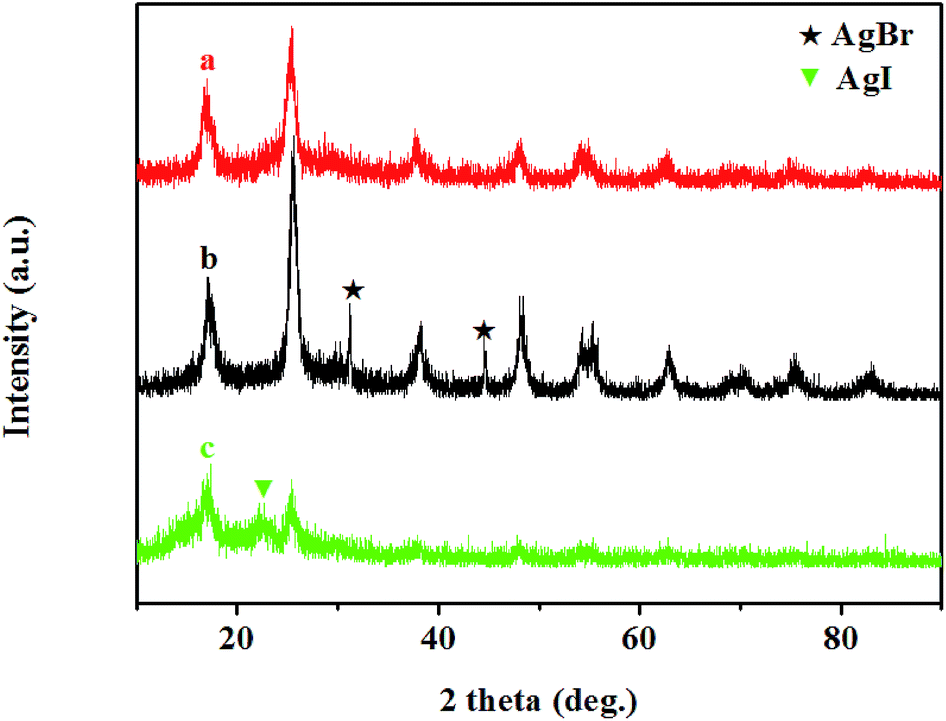 | ||
| Fig. 1 XRD patterns of (a) TiO2/PAN, (b) AgBr (10%)–TiO2/PAN and (c) AgI (10%)–TiO2/PAN. | ||
The microstructures of the obtained samples were examined by field-emission scanning electron microscopy (FE-SEM). Fig. 2a shows the FE-SEM image of PAN nanofibers, which served as supporters in the as-prepared photocatalysts. It could be clearly seen that PAN nanofibers possessed a smooth surface and a uniform diameter. As observed in Fig. 2b, TiO2/PAN hybrid nanofibers still maintained the morphology and structure of the original PAN nanofibers after solvothermal treatment. Compared with pure PAN nanofibers, uniform TiO2 nanoparticles were compactly grown on the surface of PAN nanofibers. In consequence, Ag+ ions were coupled with TiO2 nanoparticles rather than PAN nanofibers via physical adsorption process. AgX (X = Br, I) nanoparticles were generated without aggregation via gas/solid reaction (shown in Fig. 2c and d). It could be presumed that AgX nanoparticles had an intimate contact with TiO2.
 | ||
| Fig. 2 FE-SEM images of (a) PAN nanofibers, (b) TiO2/PAN, (c) AgBr (10%)–TiO2/PAN and (d) AgI (10%)–TiO2/PAN. | ||
The morphologies of the obtained samples were determined by TEM images. The low resolution TEM image of TiO2/PAN was presented in Fig. 3a. Small nanoparticles were uniformly distributed on the surface of PAN nanofibers, while the interplanar spacing of 0.352 nm corresponded to TiO2 (101) (Fig. 3b),40,42 indicating the formation of anatase TiO2 after solvothermal treatment. As shown in Fig. 3c and d, after modified by AgX (X = Br, I) nanoparticles, no obvious aggregation was found in the AgX–TiO2/PAN samples. The TEM images (Fig. 3e and f) exhibited the existence of AgX and the HRTEM images (Fig. 3g and h) further confirmed the formation of the heterojunctions between AgX and TiO2. STEM-HADDP pictures were also used to clarify the distribution of different elements in AgX–TiO2/PAN nanocomposites (Fig. 4a and b). PAN nanofibers were mainly composed of C and N, which distribution was similar to the structures of composite catalyst. Through the location of Ti and O, it could be presumed TiO2 nanoparticles were uniformly and densely coated on the surface of PAN nanofibers. There were sporadic signs of Ag and X elements due to their low content.
 | ||
| Fig. 3 TEM (a), HRTEM (b) images of TiO2/PAN (a and b); TEM (c–f) and HRTEM (g and h) images of AgBr (10%)–TiO2/PAN (c, e and g) and AgI (10%)–TiO2/PAN (d, f and h). | ||
 | ||
| Fig. 4 STEM-HAADF images of the as-prepared AgBr (10%)–TiO2/PAN (a) and AgI (10%)–TiO2/PAN (b). | ||
Fig. 5 gives the diffuse reflectance UV-vis spectra (DRS) of different photocatalysts. As shown in Fig. 5a and b, two absorption bands presented in the range of 200–300 nm attributing to the contribution of PAN polymer, while a visible-light absorption band could be observed after loading AgX on the surface of PAN nanofibers, especially for AgI, due to the light adsorption of AgX. Fig. 4c exhibits a strong adsorption peak below 390 nm, which should be attributed to the band gap energy of anatase TiO2 (3.2 eV).18,44 Except for the adsorption peak of TiO2, the as-prepared AgBr–TiO2/PAN had a little absorption in the visible light region.18,42–44 Moreover, AgI–TiO2/PAN exhibited a strong absorption band around 400–436 nm, which should be assigned to the presence of AgI.16,19,20 The diffuse reflectance UV-vis spectra future demonstrated the formation of AgX (X = Br, I).
 | ||
| Fig. 5 UV-vis diffuse reflectance spectra of (a) PAN nanofibers, (b) AgX/PAN, (c) TiO2/PAN and (d)–(h) AgX (y%)–TiO2/PAN (y = 2, 4, 6, 8, 10). | ||
The FTIR spectra of pure PAN, TiO2/PAN, AgBr (10%)–TiO2/PAN and AgI (10%)–TiO2/PAN are shown in Fig. 6. The characteristic absorption peaks at 2244 cm−1 were attributed to the stretching vibration of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N– in PAN.39,41 The peak at 1740 cm−1 might originate from the vibration of C
N– in PAN.39,41 The peak at 1740 cm−1 might originate from the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds existed in the hydrolyzed PAN nanofibers or residual DMF.39 In comparison to pure PAN, the FTIR spectra of TiO2/PAN (Fig. 6b) exhibited a broad absorption band below 1000 cm−1 corresponding to Ti–O–Ti vibration, which proved the formation of TiO2.42 In addition to characteristic absorption peaks of PAN and TiO2, no different peaks were observed in AgX (10%)–TiO2/PAN. Furthermore, a shift of the –CN– vibration was not detected in AgX (10%)–TiO2/PAN, which indicated that there was no any bond formation between –CN– group in AgX and PAN.41 So, it could be concluded that AgX nanoparticles did not connect to the PAN nanofibers.
O bonds existed in the hydrolyzed PAN nanofibers or residual DMF.39 In comparison to pure PAN, the FTIR spectra of TiO2/PAN (Fig. 6b) exhibited a broad absorption band below 1000 cm−1 corresponding to Ti–O–Ti vibration, which proved the formation of TiO2.42 In addition to characteristic absorption peaks of PAN and TiO2, no different peaks were observed in AgX (10%)–TiO2/PAN. Furthermore, a shift of the –CN– vibration was not detected in AgX (10%)–TiO2/PAN, which indicated that there was no any bond formation between –CN– group in AgX and PAN.41 So, it could be concluded that AgX nanoparticles did not connect to the PAN nanofibers.
 | ||
| Fig. 6 FTIR spectra of (a) PAN nanofibers, (b) TiO2/PAN, (c) AgBr (10%)–TiO2/PAN and (d) AgI (10%)–TiO2/PAN. | ||
Visible light photocatalytic activity and two-stage photocatalytic mechanism of AgX–TiO2/PAN
The photocatalytic activities of the as-prepared AgX–TiO2/PAN were evaluated by visible-light degradation of MO aqueous solution. For comparison, the performances of pure PAN, AgX/PAN and TiO2/PAN were also investigated. The photodegradation curves of MO over the different samples as a function of visible light irradiation time were plotted in Fig. 7. The degradation rate was represented as the variation of (1 − C/C0) with irradiation time, in which C0 and C were the concentration of MO at initial time and at certain time interval, respectively. As shown in Fig. 7a, nearly 10% of MO was absorbed by pure PAN nanofibers in dark condition, while the decolorization rate of MO kept unchanged after the light was turned on. It indicated that PAN nanofibers, a supporter in the as-prepared catalysts, only had adsorption ability toward MO, which was beneficial for the degradation of MO. Compared with TiO2/PAN and AgX/PAN (shown in Fig. 7b and c), AgX–TiO2/PAN catalysts exhibited the much higher photocatalytic activity. As shown in Fig. 7d–h, the molar ratios of AgX/TiO2 had a significant impact on the photodegradation activity. In the as-prepared AgX–TiO2/PAN system, when the AgX content increased, the AgX nanoparticles were grown dispersedly on the surface of TiO2, which was beneficial for the light adsorption. Moreover, the effective contacted interface area between AgX and TiO2 increased as the AgX ratio rose, promoting the separation of electron–hole pairs.47 Thus, the photocatalytic degradation efficiency of MO increased with the AgX loading. As shown in Fig. 7h, most of MO had been removed in the presence of AgX (10%)–TiO2/PAN after illumination for 4.5 h.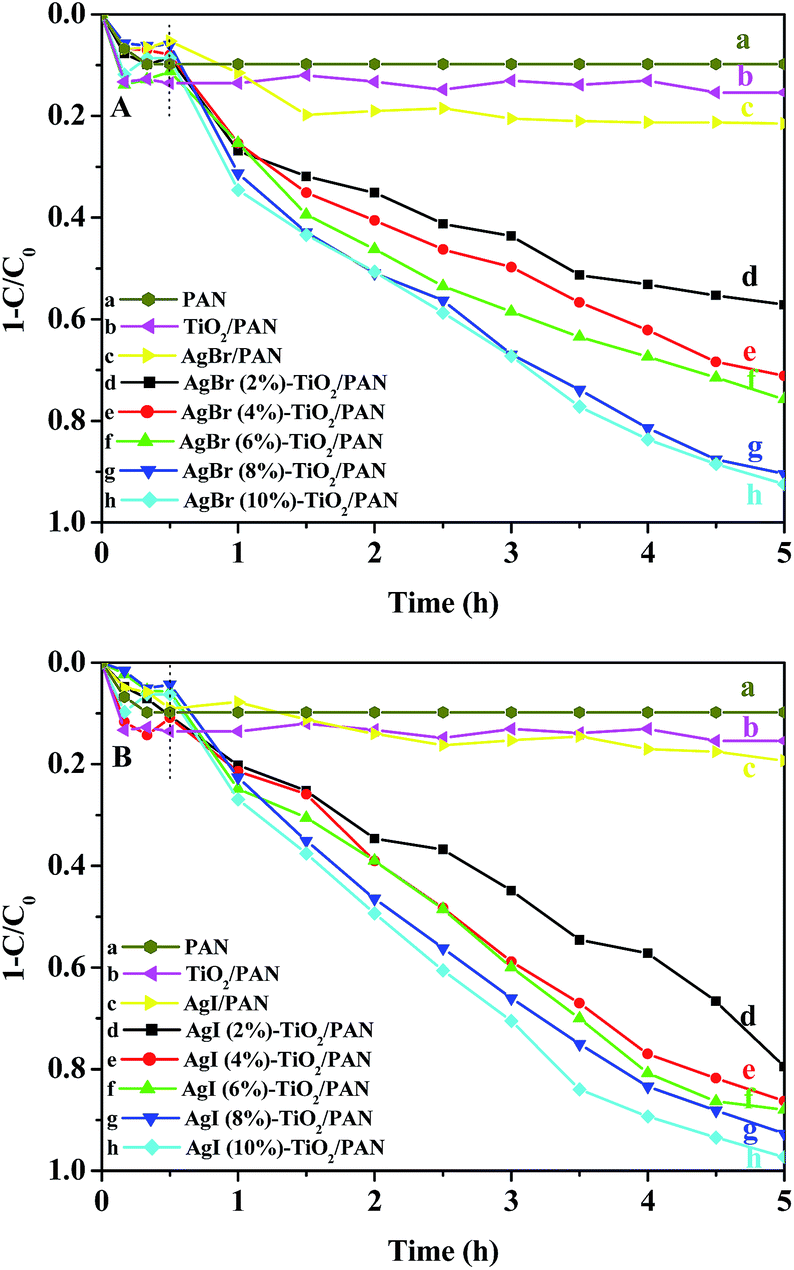 | ||
| Fig. 7 The degradation curves of MO over (a) PAN, (b) TiO2/PAN, (c) AgX/PAN and (d)–(h) AgX (y%)–TiO2/PAN (y = 2, 4, 6, 8, 10). | ||
To confirm the universal degradation ability of the as-prepared catalysts, the photocatalytic activities toward different organics were evaluated in the presence of AgBr (10%)–TiO2/PAN and AgI (10%)–TiO2/PAN. The degradation curves against irradiation time were plotted in Fig. 8A and B. The initial concentrations of methylene blue, sodium fluorescein, phenol, xylenol orange and acid red 18 were 20 mg l−1, 20 mg l−1, 20 mg l−1, 30 mg l−1 and 5 mg l−1, respectively. It was clear that the adsorption ability and degradation efficiency of the as-prepared AgX (10%)–TiO2/PAN varied with different organics and AgI (10%)–TiO2/PAN showed much higher photocatalytic activity than AgBr (10%)–TiO2/PAN for the same organic. Except for phenol solution, the color of organic pollutants gradually diminished as the irradiation time increased, suggesting that the chromophoric groups had been destroyed. It could be presumed that continuing to extend the illumination time, the organics, such as phenol and xylenol orange, could be degraded completely. In summary, the as-prepared AgX–TiO2/PAN catalysts exhibited the excellent photocatalytic activities for the degradation of methylene blue, sodium fluorescein, phenol, xylenol orange and acid red 18, which could extend their application in the field of degrading different organic pollutants in waste water.
 | ||
| Fig. 8 The adsorption and photodegradation of different organics (100 ml) in aqueous solutions containing 0.2 g AgBr (10%)–TiO2/PAN (A) or AgI (10%)–TiO2/PAN (B). | ||
Recycling experiments were carried out to evaluate the stability and durability of AgX (10%)–TiO2/PAN for MO degradation. Notably, the catalysts could be easily separated from an aqueous solution without any loss due to the application of membranous PAN. In each cycle, the adsorption process was conducted in dark condition for 30 min to establish adsorption–desorption equilibrium and then the irradiation process was lasted for 4.5 h. After 4.5 h of visible light irradiation, the catalysts were filtered, washed with distilled water and dried at 80 °C for 1 h. The recycled tests were performed for five times and described in Fig. 9. A slightly reduction of photocatalytic activity could be observed after the second run, while the degradation efficiency of MO was reduced obviously in the fourth and fifth runs, which was attributed to the photocorrosion of AgX. The decomposition of AgX could be proved by the diffuse reflectance UV-vis spectra (shown in Fig. 10b), in which a strong adsorption band appeared in visible region was attributed to the surface plasma resonance (SPR) of Ag nanoparticles.15 The catalysts recycled for five times were put in I2 (or Br2) atmosphere for regeneration, which could be convinced by the UV-vis DRS of the regenerated AgX–TiO2/PAN (Fig. 10c). Additional experiments showed that the regenerated catalysts regain high photocatalytic performance. These results demonstrated that the as-prepared AgX–TiO2/PAN catalysts had a certain photochemical stability and were easily regenerated.
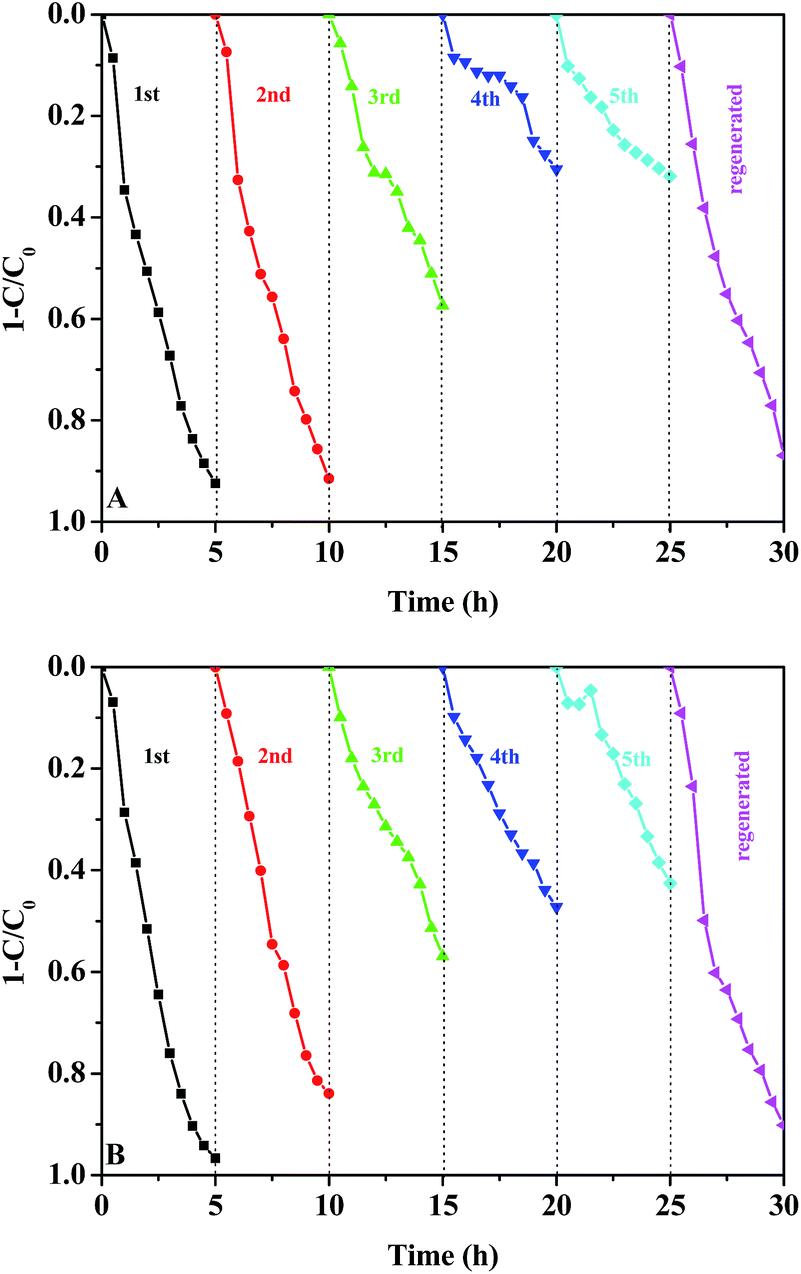 | ||
| Fig. 9 The cycling degradation efficiencies for MO of 0.2 g AgBr (10%)–TiO2/PAN (A) or AgI (10%)–TiO2/PAN (B) under visible-light irradiation. | ||
 | ||
| Fig. 10 The UV-vis diffuse reflectance spectra of (a) fresh AgX (10%)–TiO2/PAN, (b) AgX (10%)–TiO2/PAN (recycled for five runs) and (c) AgX (10%)–TiO2/PAN (regenerated). | ||
To illustrate the photocatalytic mechanism of AgX–TiO2/PAN (X = Br, I), a series of control experiments with different scavengers were carried out to investigate the generation and contribution of reactive species such as h+, ˙OH, ˙O2− and 1O2, during visible light photocatalysis. In this study, 2 mM IPA was added to quench ˙OH in the solution,16,39 0.2 mM Na2EDTA for h+,47 0.2 mM BQ for ˙O2−42,43,46,47 and 0.2 mM NaN3 for 1O2.42,43Fig. 11A and B show the photocatalytic degradation curves of MO over AgBr (10%)–TiO2/PAN and AgI (10%)–TiO2/PAN in the presence of different scavengers. The addition of NaN3 and IPA scavengers had a slightly reduction in MO degradation efficiency, which implied that 1O2 and ˙OH were not the main reactive species for MO degradation. The most depressed degradation rate occurred in the presence of Na2EDTA, indicating that h+ was the most significant reactive species. The presence of BQ also dramatically inhibited the removal efficiency of MO, which suggested that ˙O2− played an important role in the degradation of MO.
 | ||
| Fig. 11 Photodegradation curves of MO over 0.2 g AgBr (10%)–TiO2/PAN (A) or AgI (10%)–TiO2/PAN (B) under visible light irradiation without and with the presence of different scavengers. | ||
On the basis of above experimental results and related researches, the photodegradation mechanism of AgX–TiO2/PAN (X = Br, I) under visible light irradiation was proposed and illustrated in Scheme 2. The photocatalytic process could be divided into two stages for the decomposition of AgX. At the first stage, under visible light irradiation, AgX nanoparticles could be excited to produce photoinduced electrons (e−) and holes (h+), due to their narrow band gap (AgBr, 2.6 eV; AgI, 2.8 eV).43,48 The generated e− in AgX could migrate to the conduction band (CB) of TiO2 for the less negative CB of TiO2 as compared to that of AgX.48,49 Then the electrons would be trapped by surface absorbed O2 to form ˙O2− and reactive oxygen species such as ˙OH and 1O2 could be further generated. The left holes (h+) might transfer to the interface between AgX and TiO2 and oxidize X− to X0. X0 could oxidize organic pollutants while being reduced to X−. Meanwhile, ˙O2−, ˙OH and 1O2 also had a strong oxidizing ability to degrade organic pollutants. Therefore, the efficient separation of electron–hole pairs was achieved, which facilitated the photocatalytic degradation of organics. The relevant reactions could be shown in Table 1.
 | ||
| Scheme 2 Schematic illustration of two-stage photocatalytic mechanism within AgX–TiO2/PAN under visible light irradiation. | ||
| Reactions | No. |
|---|---|
| AgX + hv → AgX (e− + h+) | (1) |
| AgX (e− + h+) + TiO2 → AgX (h+) + TiO2 (e−) | (2) |
| TiO2 (e−) + O2 → ˙O2− + TiO2 | (3) |
| ˙O2− + H+ → HO2˙ | (4) |
| HO2˙ + TiO2 (e−) + H+ → H2O2 + TiO2 | (5) |
| H2O2 + TiO2 (e−) → ˙OH + OH− + TiO2 | (6) |
| ˙O2− + ˙OH → 1O2 + OH− | (7) |
| AgX (h+) + X− → AgX + X0 | (8) |
| X0 + organics → products + X− | (9) |
| ˙O2−, ˙OH, 1O2 + organics → products | (10) |
As the illumination time extended, AgX could be decomposed into Ag0. Therefore, fresh AgX–TiO2/PAN was turned into Ag–AgX–TiO2/PAN. In the newly constructed system, the electron–hole pairs could also be formed in Ag0 owing to their surface plasmon resonance (SPR) under visible light irradiation.42,43,45,46 Considering the SPR-induced local electromagnetic field and the polarization effect of negatively charged AgX surface, the photoinduced electrons and holes from Ag0 would migrate to the completely different directions. The photoinduced electrons in Ag0 might migrate to the conduction band of TiO2, while the holes would transfer from Ag0 to valance band of AgX particles, leading to the efficient separation of electron–hole pairs in the Ag0.42,43,50 Besides, the electrons generated from AgX could be injected into Ag0 and immediately migrate to the CB of TiO2.42,43 The electrons transferred from AgX could significantly inhibit the reaction where Ag+ ions of AgX might capture the electrons generated in the CB of AgX, resulting in the decomposition of AgX.43 The generation process of reactive species in this system was similar to that of fresh AgX–TiO2/PAN. The major reaction steps were summarized in Table 2. As for the regenerated catalysts, charge transfer in photocatalytic process would go through the same procedure occurred in fresh AgX–TiO2/PAN.
| Reactions | No. |
|---|---|
| AgX + hv → AgX (e− + h+) | (1) |
| Ag0 + hv → Ag* | (2) |
| Ag* + hv → TiO2 (e−) + ˙Ag+ | (3) |
| ˙Ag+ + AgX → Ag0 + AgX (h+) | (4) |
| AgX (h+) + X− → AgX + X0 | (5) |
| X0 + organics → products + X− | (6) |
| TiO2 (e−) + O2 → ˙O2− + TiO2 | (7) |
| ˙O2− + H+ → HO2˙ | (8) |
| HO2˙ + TiO2 (e−) + H+ → H2O2 + TiO2 | (9) |
| H2O2 + TiO2 (e−) → ˙OH + OH− + TiO2 | (10) |
| ˙O2− + ˙OH → 1O2 + OH− | (11) |
| ˙O2−, ˙OH, 1O2 + organics → products | (12) |
Conclusions
Novel visible light photocatalysts AgX–TiO2/PAN (X = Br, I) with different molar ratios between AgX and TiO2 were synthesized via a facile electrospinning technique, solvothermal synthesis, physical adsorption and gas/solid reaction. In the as-prepared catalysts, TiO2 nanoparticles were densely and uniformly distributed on the surface of PAN nanofibers and AgX nanoparticles were closely combined with TiO2 rather than PAN nanofibers. The fabricated catalysts exhibited a highly visible light photocatalytic performance in degrading different organics. But most of all, they could be easily separated and regenerated. Based on the experimental results, a possible photocatalytic process was also proposed. It could be concluded that the high photocatalytic activity of AgX–TiO2/PAN was attributed to the well contact between AgX and TiO2 and the efficient separation of photogenerated electron–hole pairs. The excellent photocatalytic performance and reusable property of AgX–TiO2/PAN enabled it to be a promising material in the field of environmental remediation.Acknowledgements
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (no. 21266016) and Specialized Research Fund for the Doctoral Program of Higher Education (no. 20121514120004).References
- S. Q. Liu, Z.-R. Tang, Y. G. Sun, J. C. Colmenares and Y.-J. Xu, Chem. Soc. Rev., 2015, 44, 5053 RSC.
- Z. F. Bian, T. Tachikawa, P. Zhang, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 458 CrossRef CAS PubMed.
- W.-S. Wang, H. Du, R.-X. Wang, T. Wen and A.-W. Xu, Nanoscale, 2013, 5, 3315 RSC.
- N. Zhang, M.-Q. Yang, S. Q. Liu, Y. G. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307 CrossRef CAS PubMed.
- H. F. Cheng, B. B. Huang, Y. Dai, X. Y. Qin and X. Y. Zhang, Langmuir, 2010, 26(9), 6618 CrossRef CAS PubMed.
- C. H. An, W. Jiang, J. Z. Wang, S. T. Wang, Z. H. Ma and Y. P. Li, Dalton Trans., 2013, 42, 8796 RSC.
- X. Liu, Y. X. Li, S. Q. Peng, G. X. Lu and S. B. Li, Photochem. Photobiol. Sci., 2013, 12, 1903 CAS.
- D. Dvoranová, V. Brezová, M. Mazúr and M. A. Malati, Appl. Catal., B, 2002, 37, 91 CrossRef.
- C. Han, M.-Q. Yang, N. Zhang and Y.-J. Xu, J. Mater. Chem. A, 2014, 2, 19156 CAS.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287 CrossRef CAS PubMed.
- H. Wang, T. T. You, W. W. Shi, J. H. Li and L. Guo, J. Phys. Chem. C, 2012, 116, 6490 CAS.
- W.-T. Sun, Y. Yu, H.-Y. Pan, X.-F. Gao, Q. Chen and L.-M. Peng, J. Am. Chem. Soc., 2008, 130, 1124 CrossRef CAS PubMed.
- H. Wang, J. T. Yang, X. L. Li, H. Z. Zhang, J. H. Li and L. Guo, Small, 2012, 8(18), 2802 CrossRef CAS PubMed.
- H. Xu, J. Yan, Y. G. Xu, Y. H. Song, H. M. Li, J. X. Xia, C. J. Huang and H. L. Wan, Appl. Catal., B, 2013, 129, 182 CrossRef CAS PubMed.
- P. Wang, B. B. Huang, X. Y. Zhang, X. Y. Qin, H. Jin, Y. Dai, Z. Y. Wang, J. Y. Wei, J. Zhan, S. Y. Wang, J. P. Wang and M.-H. Whangbo, Chem.–Eur. J., 2009, 15, 1821 CrossRef CAS PubMed.
- C. Hu, X. X. Hu, L. S. Wang, J. H. Qu and A. M. Wang, Environ. Sci. Technol., 2006, 40, 7903 CrossRef CAS.
- P. W. Huo, Y. S. Yan, S. T. Li, H. M. Li and W. H. Huang, Desalination, 2010, 256, 196 CrossRef CAS PubMed.
- Q. Y. Li, Y. Y. Xing, R. Li, L. L. Zong, X. D. Wang and J. J. Yang, RSC Adv., 2012, 2, 9781 RSC.
- D. Y. Wu and M. C. Long, ACS Appl. Mater. Interfaces, 2011, 3, 4770 CAS.
- Y. Z. Li, H. Zhang, Z. M. Guo, J. J. Han, X. J. Zhao, Q. N. Zhao and S.-J. Kim, Langmuir, 2008, 24, 8351 CrossRef CAS PubMed.
- W. Sun, Y. Z. Li, W. Q. Shi, X. J. Zhao and P. F. Fang, J. Mater. Chem., 2011, 21, 9263 RSC.
- C. Hu, J. Guo, J. H. Qu and X. X. Hu, Langmuir, 2007, 23, 4982 CrossRef CAS PubMed.
- M. Pirhashemi and A. Habibi-Yangjeh, J. Alloys Compd., 2014, 601, 1 CrossRef CAS PubMed.
- J. Li, W. W. Tu, H. B. Li, J. C. Bao and Z. H. Dai, Chem. Commun., 2014, 50, 2108 RSC.
- K. Vignesh, A. Suganthi, M. Rajarajan and S. A. Sara, Powder Technol., 2012, 224, 331 CrossRef CAS PubMed.
- L. Q. Ye, J. Y. Liu, C. Q. Gong, L. H. Tian, T. Y. Peng and L. Zan, ACS Catal., 2012, 2, 1677 CrossRef CAS.
- L. Kong, Z. Jiang, H. H. Lai, R. J. Nicholls, T. C. Xiao, M. O. Jones and P. P. Edwards, J. Catal., 2012, 293, 116 CrossRef CAS PubMed.
- C. Prahsarn, W. Klinsukhon and N. Roungpaisan, Mater. Lett., 2011, 65, 2498 CrossRef CAS PubMed.
- C. Y. Su, Y. F. Tong, M. Y. Zhang, Y. Zhang and C. L. Shao, RSC Adv., 2013, 3, 7503 RSC.
- Y. Y. Liang, H. L. Wang, H. S. Casalongue, Z. Chen and H. J. Dai, Nano Res., 2010, 3, 701 CrossRef CAS.
- S. Izadyar and S. Fatemi, Ind. Eng. Chem. Res., 2013, 52, 10961 CrossRef CAS.
- H.-W. Liang, W.-J. Zhang, Y.-N. Ma, X. Cao, Q.-F. Guan, W.-P. Xu and S.-H. Yu, ACS Nano, 2011, 5(10), 8148 CrossRef CAS PubMed.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233 CrossRef CAS PubMed.
- Y. H. Zhang, Z.-R. Tang, X. Z. Fu and Y.-J. Xu, ACS Nano, 2010, 4(12), 7303 CrossRef CAS PubMed.
- C. Y. Su, X. Ran, J. L. Hu and C. L. Shao, Environ. Sci. Technol., 2013, 47, 11562 CrossRef CAS PubMed.
- H. C. Yu, Q. S. Dong, Z. B. Jiao, T. Wang, J. T. Ma, G. X. Lu and Y. P. Bi, J. Mater. Chem. A, 2014, 2, 1668 CAS.
- S. K. Nataraj, K. S. Yang and T. M. Aminabhavi, Prog. Polym. Sci., 2012, 37, 487 CrossRef CAS PubMed.
- J. Bai, Y. X. Li, S. T. Yang, J. S. Du, S. G. Wang, C. Q. Zhang, Q. B. Yang and X. S. Chen, Nanotechnology, 2007, 18, 305601 CrossRef.
- Y. C. Chou, C. L. Shao, X. H. Li, C. Y. Su, H. C. Xu, M. Y. Zhang, P. Zhang, X. Zhang and Y. C. Liu, Appl. Surf. Sci., 2013, 285P, 509–516 CrossRef PubMed.
- X. P. Wang and T.-T. Lim, Water Res., 2013, 47, 4148 CrossRef CAS PubMed.
- G. Panthi, S.-J. Park, T.-W. Kim, H.-J. Chung, S.-T. Hong, M. Park and H.-Y. Kim, J. Mater. Sci., 2015, 50, 4477 CrossRef CAS.
- P. H. Wang, Y. X. Tang, Z. L. Dong, Z. Chen and T.-T. Lim, J. Mater. Chem. A, 2013, 1, 4718 CAS.
- X. P. Wang, Y. X. Tang, Z. Chen and T.-T. Lim, J. Mater. Chem., 2012, 22, 23149 RSC.
- C. Hu, Y. Q. Lan, J. H. Qu, X. X. Hu and A. M. Wang, J. Phys. Chem. B, 2006, 110, 4066 CrossRef CAS PubMed.
- J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394 CAS.
- B. Z. Tian, R. F. Dong, J. M. Zhang, S. Y. Bao, F. Yang and J. L. Zhang, Appl. Catal., B, 2014, 158–159, 76 CrossRef CAS PubMed.
- L. Yin, Z. Wang, L. Lu, X. K. Wan and H. X. Shi, New J. Chem., 2015, 39, 4891 RSC.
- J. H. Yi, L. L. Huang, H. J. Wang, H. Yu and F. Peng, J. Hazard. Mater., 2015, 284, 207 CrossRef CAS PubMed.
- M. A. Asi, C. He, M. H. Su, D. H. Xia, L. Lin, H. Q. Deng, Y. Xiong, R. L. Qiu and X.-Z. Li, Catal. Today, 2011, 175, 256 CrossRef PubMed.
- D. D. Yu, J. Bai, H. O. Liang, J. Z. Wang and C. P. Li, Appl. Surf. Sci., 2015, 349, 241 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |