
Prediction of stoichiometric PoHn compounds: crystal structures and properties†
Yunxian Liu,
Defang Duan,
Fubo Tian,
Chao Wang,
Gang Wu,
Yanbin Ma,
Hongyu Yu,
Da Li ,
Bingbing Liu and
Tian Cui*
,
Bingbing Liu and
Tian Cui*
State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, People's Republic of China. E-mail: cuitian@jlu.edu.cn
First published on 30th November 2015
Abstract
Stable compounds, crystal structures and properties of polonium hydrides have been systematically investigated through the first-principles calculations based on the density functional theory. With the increasing pressure, several stoichiometries (PoH, PoH2, PoH4 and PoH6) are predicted to be stabilized in an excess hydrogen environment. Except for PoH, other stable stoichiometries exhibit intriguing structural character with the appearance of H2 units. Moreover, the electronic band structure and projected density of states (PDOS) demonstrate that these energetically stable phases are metallic. The application of the Allen–Dynes modified McMillan equation with the calculated electron-phonon coupling parameter reveals that PoH4 is a superconductor with a critical temperature Tc of 41.2–47.2 K at 200 GPa.
1. Introduction
Over the past ten years, hydrogen-rich compounds have been the focus of research for their potential to become extraordinary high-temperature superconductors at lower pressures which are accessible to current experiments. The elements of group VIA, which include oxygen, sulfur, selenium, tellurium and polonium, present many interesting properties under pressure, such as the metallic phases of O2, S, Se and Te were found to be superconducting in experiment, with a transition temperature Tc of 0.6, 10, 5 and 4.5 K, respectively.1–5 Recently, group VIA hydrides were expected to metalize at lower pressures with potential superconductivity. Remarkably, the Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m phase of H3S with outstanding high Tc of 191–204 K at 200 GPa was first predicted by our group using the ab initio calculation method.6 It is spectacular that the superconductivity in H2S sample with Tc of 190 K above 150 GPa was observed by Eremets et al. in experiment,7 which is speculated that the high Tc likely associates with our predicted H3S. Later, we theoretically reported that H2S really decomposes into S and H3S above 43 GPa, and H3S is stable at least up to 300 GPa, which proved that high Tc in H2S sample observed experimentally comes from H3S.8 Other theoretical studies also confirmed our results using different structural search techniques.9,10 Recently, selenium (Se) hydrides and tellurium (Te) hydrides were theoretically predicted to exhibit Tc in the range of 40–131 K and 46–104 K at megabar pressures, respectively.11–13 These dense hydrides has reignited great interest in chalcogen hydrogen-rich compounds.
m phase of H3S with outstanding high Tc of 191–204 K at 200 GPa was first predicted by our group using the ab initio calculation method.6 It is spectacular that the superconductivity in H2S sample with Tc of 190 K above 150 GPa was observed by Eremets et al. in experiment,7 which is speculated that the high Tc likely associates with our predicted H3S. Later, we theoretically reported that H2S really decomposes into S and H3S above 43 GPa, and H3S is stable at least up to 300 GPa, which proved that high Tc in H2S sample observed experimentally comes from H3S.8 Other theoretical studies also confirmed our results using different structural search techniques.9,10 Recently, selenium (Se) hydrides and tellurium (Te) hydrides were theoretically predicted to exhibit Tc in the range of 40–131 K and 46–104 K at megabar pressures, respectively.11–13 These dense hydrides has reignited great interest in chalcogen hydrogen-rich compounds.
Polonium (Po) is the only metal element in the group VIA, which possesses the simple cubic (sc) structure at ambient conditions. Based on the high-Tc superconductivity of other chalcogenide hydrides, polonium hydrides might also exhibit high superconducting transition temperature at high pressures. It is well established that the application of high pressure can influence the physical and chemical properties of matter, and lead to the formation of unprecedented phases or complexes with novel characters. Such as, the superconductivity of SiH4 and PtH were experimentally confirmed at high pressure.14–16 Other new unusual stoichiometries were predicted to form at high pressure in theory (e.g. LiH6, NaH9, KH6, RbH5, CsH3 and so on).17–21 Therefore, the investigation of polonium hydrides under high pressure might provide us interesting information.
In this article, the ground-state structures of Po–H system over a range of pressures were explored by using the recently fast-developed evolutionary algorithm Universal Structure Predictor: Evolutionary Xtallography (USPEX).22–24 Moreover, we investigate the optimum static structures as a function of pressure, their dynamical stability, the corresponding electronic band structures and superconductivity of Po–H system. Our results show that except for PoH, other stable stoichiometries have H2 units. Furthermore, all the stable polonium hydrides are found to be metallic, and the superconducting critical temperature Tc values of PoH, PoH4, and PoH6 are 0.14–0.65 K, 41.1–47.2 K, and 2.65–4.68 K at 300, 200 and 200 GPa, respectively.
2. Computational method
The searches for stable high pressure structures of the PoHn (n = 1–6) system were performed through the evolutionary algorithm, implemented in the USPEX code.22–24 Structure predictions were implemented at 50, 100, 200, and 300 GPa with 1–4 formula units (f.u.). Each structure was fully relaxed to an energy minimum using density functional theory with the Perdew–Burke–Ernzerhof (PBE) form of the generalized gradient approximation (GGA)25 implemented in the Vienna ab initio simulation package VASP code.26 The hybrid functional of Heyd, Scuseria, and Ernzerhof (HSE06)27,28 was also used to verify the results of band structures with GGA function. We optimized predicted stable structures at higher accurate level. A plane-wave basis set cutoff of 800 eV and a Brillouin zone sampling grid of spacing 2π × 0.03 Å−1 were chose, which ensures total energy convergence better than 1 meV per atom. The all-electron projector augmented wave method (PAW)29 is adopted with the cut-off radius of 0.8 a.u. and 2.1 a.u. for H (1s2) and Po (6s26p4), respectively. In the geometrical optimization, all forces on atoms were converged to less than 0.005 eV Å−1. The lattice dynamics were performed using the supercell approach30 as implemented in PHONOPY program.31 The electron–phonon coupling (EPC) for superconducting properties of stable compounds have been computed with QUANTUM-ESPRESSO.32 Convergence tests provide a suitable value of 80 Ry kinetic energy cutoff. The q-point mesh in the first BZ of 4 × 4 × 2 for P63/mmc (PoH), 5 × 5 × 2 for C2/c (PoH4), and 3 × 3 × 3 for C2/m (PoH6) structures were used in the Brillouin zone.3. Results and discussion
We explored polonium hydrides with hydrogen-rich contents through the evolutionary algorithm USPEX. The calculations were performed at 50–300 GPa with considering simulation sizes ranging from one to four formula units (f.u.) for PoHn (n = 1, 2, 3, 4, 5, 6). Our calculations give convex hulls on the formation enthalpy of the Po–H system with H-rich stoichiometry from 50 to 300 GPa, as shown in Fig. 1. The structures on the convex hull are thermodynamically stable and they may be experimentally synthesized in principle, whereas those above it are metastable. The essential information can be summarized as follows: (i) at 100 GPa, PoH2 has the most negative enthalpy out of all the structures that we examined; which can be synthesized using Po and H2 by the reaction Po + H2 → PoH2; (ii) when pressure is increased up to 150 GPa, PoH2 is still the global minimum stoichiometry. And PoH4 also falls on the convex hull, which suggests that it might be synthesized via the reaction PoH2 + H2 → PoH4, and it keeps stable up to 300 GPa; (iii) at 200 GPa, PoH2, PoH4 and PoH6 stoichiometries are all found to lie on the hull; (vi) with increasing pressure to 250 GPa, another stable stoichiometry of PoH appears on the convex hull, which can be formed by the reaction PoH2 + Po → 2PoH2; (v) at 300 GPa, except for PoH3 and PoH5, all the stoichiometries become stable and emerge on the hull. The stable pressure ranges of predicted PoH, PoH2, PoH4 and PoH6 together with Po and H2 (ref. 33 and 34) are depicted in Fig. 2. At 84.5 GPa, the Cmcm phase of PoH2 becomes stable and remains up to 133 GPa. Then the Pnma-PoH2 becomes energetically favorable. PoH4 (space group C2/c), PoH6 (space group C2/m) and PoH (space group P63/mmc) become stable at 137, 195.5 and 208 GPa, respectively. | ||
| Fig. 1 The enthalpies of formation (ΔHf, in eV per atom) for Po–H system with respect to Po and H at 50, 100, 150, 200, 250 and 300 GPa. | ||
 | ||
| Fig. 2 The phase diagram of Po–H system in the pressure range from 70 to 300 GPa. | ||
The crystal structures motifs of stable stoichiometries are depicted in Fig. 3, which shows that all structures contain H2 units exception of PoH. We think that as the electronegativity value (2.0) of Po is smaller than H (2.2), charges transfer from Po to H. For PoH compound, the content of H is relative small, and the interaction of Po–H is enough to make the PoH stoichiometry stable. While for PoH2,4,6, the content of H is more, H atoms are tend to form H2 units to decrease material energy. Therefore, the H2 units are present in PoH2,4,6 (which contain more H), while H2 units don't appear in PoH. From our calculations, the predicted energetically favored structure of PoH is P63/mmc at 300 GPa (Fig. 3a), and it is isostructural with TeH.13 The PoH2 stoichiometry has two structures with Cmcm and Pnma space group at 50 and 300 GPa, respectively. To better describe the structural feature and to prevent misunderstanding, we connected the Po atoms with a purple straight line, as shown in Fig. 3b and d. While for the cases of PoH2-Pnma, PoH4-C2/c, and PoH6-C2/m structures (Fig. 3b–e), some H atoms are existent in form of H2 units and form bonds, which is due to the distances of H–H is in the range of 0.793–0.818 Å. For Cmcm structure, Po atoms form three-dimensional network (Fig. 3b), while in Pnma phase, Po atoms form a distorted bcc lattice (Fig. 3c). Moreover, quasi-molecular H2 units are found with the distance of H–H 0.793 Å (50 GPa in Cmcm) and 0.841 Å (200 GPa in Pnma), respectively. For PoH4 stoichiometry (Fig. 3d), it posses C2/c symmetry at 200 GPa, where Po atoms built up 2D layers and the H2 units have H–H distance of 0.818 Å. Turning to the richest-H2 stoichiometry PoH6, it has a monoclinic C2/m space group at 200 GPa with chain Po atoms and H2 units, as shown in Fig. 3e. By observing the high pressure structural feature for the chalcogenide group hydrides, we discovered that some hydrogen are existent in form of H2 unit in S–H,6 Se–H,11 Te–H13 and Po–H systems. Calculated structural parameters of the predicted stable structures for Po–H compounds at the selected pressure are displayed in Table S1.† It is necessary to determine the dynamical stability of structures. Fig. 4 presents the calculated phonon band dispersions and projected phonon density of states (PHDOS) for PoH-P63/mmc, PoH2-Pnma, PoH4-C2/c, and PoH6-C2/m. They are all dynamical stability, as there are no imaginary phonon mode in the entire Brillouin zone. In addition, we see that there exist a distinguish between PoH and PoH2,4,6, which is attributed to that PoH2,4,6 posses H2 units. For the former (Fig. 4a), the phonon band dispersions vibrational modes can be divided into two parts, low energy phonon modes are mainly from Po atoms, whereas modes with high frequency region are associated with H atoms. For the later (Fig. 4b–d), the vibrational modes can be separate into three regions. The low-energy region, the intermediate-frequency region and high frequencies are dominated by Po atoms, H⋯Po⋯H vibrations and H2 units, respectively.
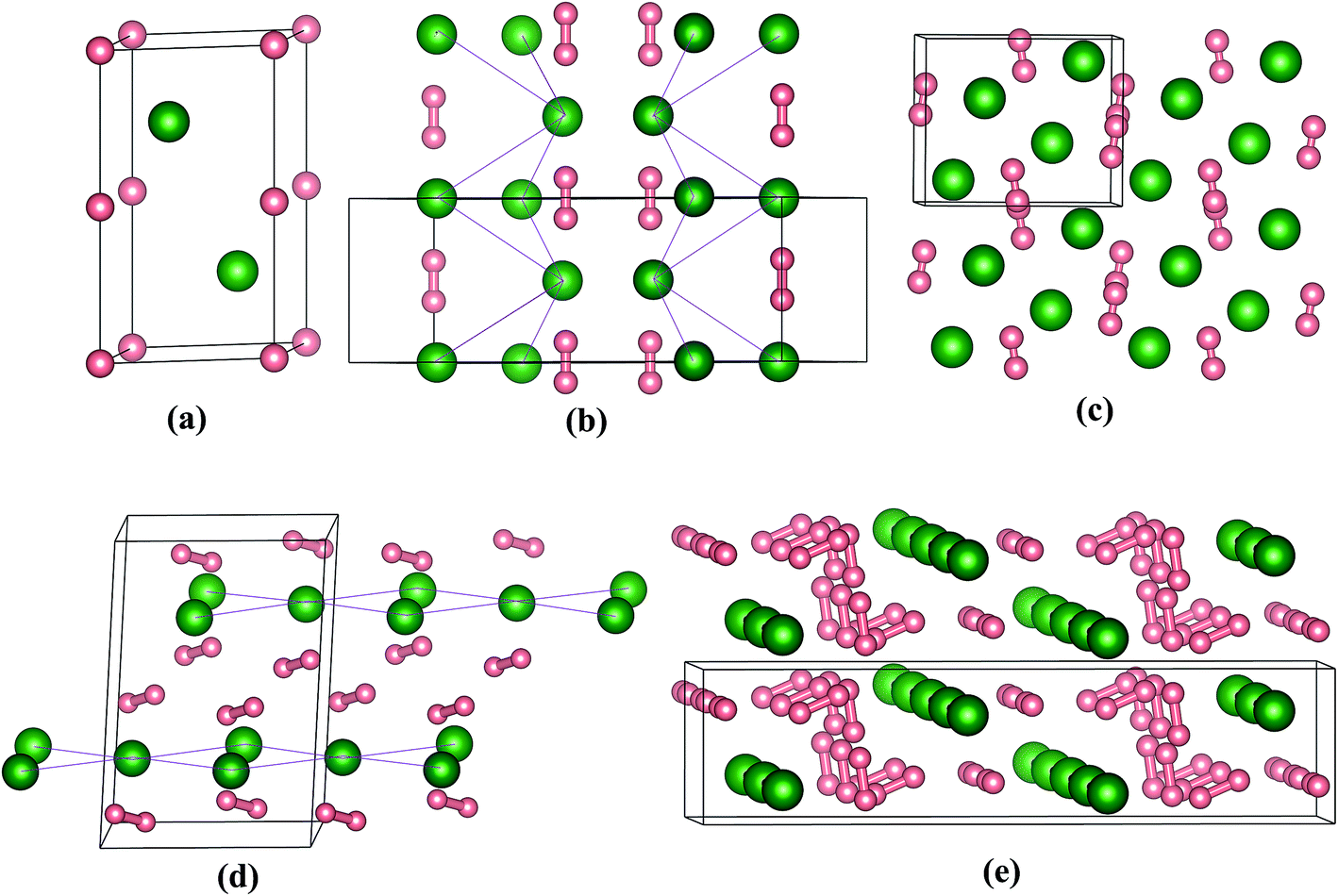 | ||
| Fig. 3 The selected stable phases for Po–H system. Green atoms depict Po, while pink atoms present H, (a) PoH-P63/mmc at 300 GPa, (b) PoH2-Cmcm at 50 GPa, (c) PoH2-Pnma at 300 GPa, (d) PoH4-C2/c at 200 GPa and (e) PoH6-C2/m at 200 GPa. | ||
 | ||
| Fig. 4 (a–d) The phonon band structure and projected phonon DOS charts for PoH-P63/mmc, PoH2-Pnma, PoH4-C2/c and PoH6-C2/m at different pressure, respectively. | ||
Band structure and projected density of states (DOS) at different pressures for PoH, PoH2, PoH4 and PoH6 in the P63/mmc, Pnma, C2/c and C2/m phases were calculated, as displayed in Fig. 5. We clearly see that all the above structures exhibit metallic feature with overlap between the conduction and valence bands. For the simplest stoichiometry PoH-P63/mmc structure and PoH2-Pnma phase, the Po-p state predominates the energy range from −10 to 5 eV, and there exist strong hybridization between Po-p and H-s states (−10 to −15 eV), as can be seen in Fig. 5a and b. For PoH4, we can see that Po-s, Po-p, and H-s orbits dominate the Fermi level (Fig. 5c). For H-rich C2/m of PoH6, at the Fermi level a strong DOS peak is observed in Fig. 5d, which originates from Po-p and H-s electrons. Note that, at Fermi level the Po-p is mainly dominated in Po–H system, which is similar to Se–H and Te–H systems. And this same behavior is attributed to their isoelectronic nature.11,13 As we know, the GGA function underestimate the band gap, so we calculate band structures of PoH-P63/mmc and PoH2-Pnma phases with the hybrid functional HSE06 method. It is shown that the results for the GGA and HSE calculations were almost identical, except that the band structures move down in HSE06 (Fig. S1†).
 | ||
| Fig. 5 Electronic band structure and partial density of states (PDOS). (a) PoH-P63/mmc at 300 GPa, (b) PoH2-Pnma at 200 GPa, (c) PoH4-C2/c at 200 GPa and (d) PoH6-C2/m at 200 GPa. | ||
To observe the possible superconductivity of PoH-P63/mmc, PoH2-Pnma, PoH4-C2/c and PoH6-C2/m, we investigate the electron–phonon coupling (EPC) parameter λ, the logarithmic average phonon frequency ωlog, and the Eliashberg phonon spectral function α2F(ω) to explore the possible superconductivity for PoH-P63/mmc, PoH2-Pnma, PoH4-C2/c and PoH6-C2/m at different pressures. The obtained EPC parameter λ is 0.33 for PoH (300 GPa), 0.19 for PoH2 (200 GPa), 1.08 for PoH4 (200 GPa) and 0.43 for PoH6 (200 GPa), while the calculated ωlog from the phonon spectrum reach 806, 713, 603 and 836 K, respectively. The superconducting critical temperature Tc of Po–H system can be estimated by using the Allen–Dynes-modified McMillan equation35 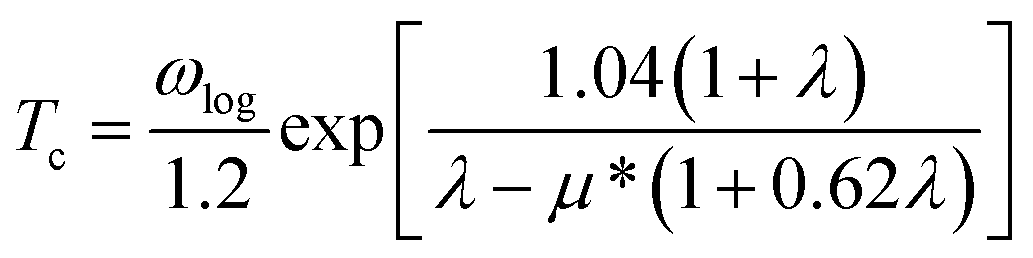 with the calculated logarithmic average frequency ωlog and the Coulomb pseudopotential μ* (0.1–0.13). The calculated Tc for PoH-P63/mmc, PoH2-Pnma, PoH4-C2/c and PoH6-C2/m are 0.14–0.65 K at 300 GPa, 0 K at 200 GPa, 41.1–47.2 K at 200 GPa and 2.25–4.68 K at 200 GPa, respectively. We also calculated Tc for PoH4-C2/c as a function of pressure. The values of ωlog, N(Ef), λ and Tc are summarized in Table S2.† As pressure increased, the resulting ωlog and Tc reach up to 804.2 K and 45.7–53.6 K at 250 GPa, respectively. However, both N(Ef) and λ parameters decrease with increasing pressure. The results indicated that the raising ωlog mainly causes the positive pressure dependence of Tc.
with the calculated logarithmic average frequency ωlog and the Coulomb pseudopotential μ* (0.1–0.13). The calculated Tc for PoH-P63/mmc, PoH2-Pnma, PoH4-C2/c and PoH6-C2/m are 0.14–0.65 K at 300 GPa, 0 K at 200 GPa, 41.1–47.2 K at 200 GPa and 2.25–4.68 K at 200 GPa, respectively. We also calculated Tc for PoH4-C2/c as a function of pressure. The values of ωlog, N(Ef), λ and Tc are summarized in Table S2.† As pressure increased, the resulting ωlog and Tc reach up to 804.2 K and 45.7–53.6 K at 250 GPa, respectively. However, both N(Ef) and λ parameters decrease with increasing pressure. The results indicated that the raising ωlog mainly causes the positive pressure dependence of Tc.
Fig. 6 shows the evaluated Eliashberg phonon spectral function α2F(ω) and the partial electron–phonon integral λ(ω) for PoH-P63/mmc, PoH4-C2/c and PoH6-C2/m to understand the origin of λ. Taken altogether, the electron–phonon integral has two regions low and high frequency in the above structures. For P63/mmc structure of PoH at 300 GPa (Fig. 6a), the low-frequency modes below 10 THz, contributing 46% of the total value λ, come from Po vibrations, whereas high frequency modes above 36 THz associated with H atoms provide a contribution 54% in total EPC parameter λ. For the C2/c phase of PoH4 at 200 GPa (Fig. 6b), Po vibrations dominate the low frequency region and contribute approximately 39.3% in total λ. The intermediate-frequency vibrational modes from H⋯Po⋯H contribute 55.1% to the λ. And the remaining 5.6% of λ is from the H2 vibrations. In the same way, for PoH6-C2/m at 200 GPa (Fig. 6c), the Po, H⋯Po⋯H and H2 vibration modes account for 25.5%, 68.3% and 7.2% of λ, respectively. Note that, in the PoH-P63/mmc which contains Po and H atom, the λ is mainly contributed from H vibrational modes. For the PoH4-C2/c and PoH6-C2/m, possessing H2 units, the low and intermediary modes are mostly constitute of the λ, while the high frequencies of H2 make small contributions to λ. The superconductive mechanism of PoH4,6 is similar to TeH4 (ref. 13) that contain similar H2 units.
 | ||
| Fig. 6 The Eliashberg phonon spectral function α2(ω) and the partial electron–phonon integration λ(ω). (a) PoH-P63/mmc at 300 GPa, (b) PoH4-C2/c at 200 GPa and (c) PoH6-C2/m at 200 GPa. | ||
4. Conclusions
In summary, we explore the structures, phase diagram and superconductivity of polonium hydrides under pressure using ab initio calculations. The polonium hydrides become thermodynamically stable with respect to decomposition into the Po and H above 84.5 GPa. Upon further compression and in the excess hydrogen environment, PoH, PoH2, PoH4 and PoH6 hydrides become thermodynamically preferred. Except for PoH-P63/mmc, a remarkable feature of the predicted stable structures is the presence of H2 units. Electron–phonon coupling calculations show that the Tc of PoH-P63/mmc, PoH4-C2/c, and PoH6-C2/m are 0.14–0.65 K at 300 GPa, 41.1–47.2 K at 200 GPa and 2.25–4.68 K at 200 GPa, respectively. Our results provide a better understanding of the pressure-induced phase diagram and superconductivity of Po–H system, which has major implications for investigating other hydrides under pressure.Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2011CB808200), National Natural Science Foundation of China (No. 51572108, 11204100, 11504127, 11504127, 11404134), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1132), National Found for Fostering Talents of basic Science (No. J1103202). China Postdoctoral Science Foundation (2012M511326, 2013T60314, and 2014M561279). Graduate Innovation Fund of Jilin University (2015102). Parts of calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.References
- K. Shimizu, K. Suhara, M. Ikumo, M. Eremets and K. Amaya, Nature, 1998, 393, 767–769 CrossRef CAS.
- F. Bundy and K. Dunn, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 3157–3164 CrossRef CAS.
- Y. Akahama, M. Kobayashi and H. Kawamura, Solid State Commun., 1992, 84, 803–806 CrossRef CAS.
- V. V. Struzhkin, R. J. Hemley, H.-K. Mao and Y. A. Timofeev, Nature, 1997, 390, 382–384 CrossRef.
- E. Gregoryanz, V. Struzhkin, R. Hemley, M. Eremets, H.-K. Mao and Y. Timofeev, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 064504 CrossRef.
- D. Duan, Y. Liu, F. Tian, D. Li, X. Huang, Z. Zhao, H. Yu, B. Liu, W. Tian and T. Cui, Sci. Rep., 2014, 4, 6968 CrossRef CAS PubMed.
- A. Drozdov, M. Eremets and I. Troyan, arXiv preprint arXiv:1412.0460, 2014.
- D. Duan, X. Huang, F. Tian, D. Li, H. Yu, Y. Liu, Y. Ma, B. Liu and T. Cui, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 180502 CrossRef.
- N. Bernstein, C. Hellberg, M. Johannes, I. Mazin and M. Mehl, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 91, 060511 CrossRef.
- I. Errea, M. Calandra, C. J. Pickard, J. Nelson, R. J. Needs, Y. Li, H. Liu, Y. Zhang, Y. Ma and F. Mauri, arXiv preprint arXiv:1502.02832, 2015.
- S. Zhang, Y. Wang, J. Zhang, H. Liu, X. Zhong, H. Sonf, L. Zhang and Y. Ma, Sci. Rep., 2015, 5, 15433 CrossRef CAS PubMed.
- J. A. Flores-Livas, A. Sanna and E. Gross, arXiv preprint arXiv:1501.06336, 2015.
- X. Zhong, Y. Wang, H. Liu, S. Zhang, G. Yang, L. Zhang and Y. Ma, arXiv preprint arXiv:1503.00396, 2015.
- M. Eremets, I. Trojan, S. Medvedev, J. Tse and Y. Yao, Science, 2008, 319, 1506–1509 CrossRef CAS PubMed.
- T. Scheler, O. Degtyareva, M. Marqués, C. L. Guillaume, J. E. Proctor, S. Evans and E. Gregoryanz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 214106 CrossRef.
- X. J. Chen, V. V. Struzhkin, Y. Song, A. F. Goncharov, M. Ahart, Z. Liu, H. K. Mao and R. J. Hemley, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20–23 CrossRef CAS PubMed.
- E. Zurek, R. Hoffmann, N. W. Ashcroft, A. R. Oganov and A. O. Lyakhov, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17640–17643 CrossRef CAS PubMed.
- P. Baettig and E. Zurek, Phys. Rev. Lett., 2011, 106, 237002 CrossRef PubMed.
- D. Zhou, X. Jin, X. Meng, G. Bao, Y. Ma, B. Liu and T. Cui, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014118 CrossRef.
- J. Hooper and E. Zurek, Chem.–Eur. J., 2012, 18, 5013–5021 CrossRef CAS PubMed.
- A. Shamp, J. Hooper and E. Zurek, Inorg. Chem., 2012, 51, 9333–9342 CrossRef CAS PubMed.
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef PubMed.
- A. R. Oganov, A. O. Lyakhov and M. Valle, Acc. Chem. Res., 2011, 44, 227–237 CrossRef CAS PubMed.
- A. O. Lyakhov, A. R. Oganov, H. T. Stokes and Q. Zhu, Comput. Phys. Commun., 2013, 184, 1172–1182 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- K. Parlinski, Z. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- A. Rubio-Ponce, J. Morales and D. Olguín, in Advances in the Theory of Quantum Systems in Chemistry and Physics, Springer, 2012, pp. 119–127 Search PubMed.
- C. J. Pickard and R. J. Needs, Nat. Phys., 2007, 3, 473–476 CrossRef CAS.
- P. B. Allen, Phys. Rev. B: Condens. Matter Mater. Phys., 1975, 12, 905–922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19223d |
| This journal is © The Royal Society of Chemistry 2015 |