
Gas separation membranes made through thermal rearrangement of ortho-methoxypolyimides†
Abstract
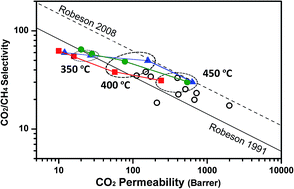
ortho-Methoxypolyimides were prepared from 3,3ʹ-dimethoxybenzidine (DMAB) and hexafluoroisopropylidene diphthalic anhydride (6FDA). High molecular weights of the resulting polymers were achieved, and the physical properties of the materials were investigated. The polyimides exhibited excellent thermal properties and film-forming capabilities. Polyimides were treated at high temperatures to promote thermal rearrangement (TR) processes. The final composition of the TR polymers seems not to correspond to TR-polybenzoxazoles, as was the case when analogous ortho-hydroxypolyimides were exposed to similar thermal treatments. A detailed study was carried out to elucidate the actual mechanism of the thermal rearrangement, comparing ortho-hydroxypolyimides, ortho-acetylpolyimides and ortho-methoxypolyimides. Results led to the conclusion that the chemical nature of the final TR polymers attained from ortho-methoxypolyimides is complex since imide, lactam and benzoxazole groups all seem to be present after thermal treatment. Gas permeation properties exhibited by thermally treated ortho-methoxypolyimides compared well with those of other TR-PBOs, showing CO2 permeability of 540 Barrers.
 Please wait while we load your content...
Please wait while we load your content...

