
[TM13@Bi20]− clusters in three-shell icosahedral matryoshka structure: being as superatoms
C. Y. Koua,
L. Zhuanga,
G. Q. Wangb,
H. Cuic,
H. K. Yuan*a,
C. L. Tiana,
J. Z. Wanga and
H. Chen*a
aSchool of Physical Science and Technology, Southwest University, Chongqing, 400715, People's Republic of China. E-mail: yhk10@swu.edu.cn; Fax: +86-023-68254608
bCollege of Arts and Science, Shanxi Aviation Professional Technical Institute, Hanzhong, 723102, People's Republic of China
cSchool of Automation and Information Engineering, Xi'an University of Technology, Xi'an 710048, People's Republic of China
First published on 22nd October 2015
Abstract
Using the density functional theory (DFT) method, the 33-atom intermetalloid [TM13@Bi20]− clusters (TM = 3d, 4d), which are composed of Bi20 pentagonal dodecahedra surrounding TM12 icosahedra with a single TM atom at the center, have been systematically examined to explore the possibility of clusters being as superatoms. The results show that most TM13 clusters can be attractively encapsulated into Bi20 cage to form a stable core–shell configuration, exhibiting an interesting progression of thermal stability along the 3d and 4d periods. Taking into account the structural stability (binding energy, embedding energy, and core–shell interaction) as well as the chemical stability (HOMO–LUMO gap), we proposed that [TM13@Bi20]− clusters with Ti and Mn doping in 3d series (Zr and Ag doping in 4d series) are specially stable and to be the protyle superatoms. For such systems, the molecular orbital shapes and energy alignments are in analogy with the atomic patterns, coinciding the general characters of superatomic orbitals. The closed core superatomic shell together with the partially-filled valence superatomic shell configuration leads to magnetic moment in stable [TM13@Bi20]−, e.g., [Mn13@Bi20]− cluster with the half-filled subshell can be assigned as a magnetic superatom owning to its modest HOMO–LUMO gap of 0.37 eV and large magnetic moment of 36 μB. The exchange-splitting in TM-3d states is found to be the driving force for the improvement of exchange-splitting of superatomic states.
1 Introduction
One of the most exciting developments in cluster science is the finding that electronic states in specific clusters can be reasonably described by a nearly confined free electron gas in a spherically symmetric potential, and their unusual stability and chemical reactivities can be understood in terms of the spherical shell jellium model.1–3 Such novel clusters, named as the superatoms,4 can mimic elemental atoms and have the potentials to construct a three-dimensional periodic table on the basis of the difference between its valence electron count and the number of electrons required to fill the nearest superatomic shell.5,6 In particular, utilizing these artificial clusters (superatoms) as the building-blocks, one could design new materials with the desired characteristics.4–8 Like free atom, superatomic electronic states are proposed to bunch into several supershells of 1S21P61D102S21F142P61G182D10…, much in the same way as 1s22s22p63s23p63d10… in free atoms but in different combination of quantum numbers and orbital sequences. Unlike free atom, the superatomic orbital (superorbital) spans over multiple atoms and the filling of orbitals does not usually exhibit Hund's rule. The essential idea of superatom is that specific stable cluster would exhibit similar chemical patterns of free atom if it has analogous valence shells with free atom.The first step toward the development of superatoms is to identify the stable motifs, understand electronic features, and control their stabilities. Since the valance electrons in simple-metal clusters can be universally treated as metallic or more precisely as a confined nearly free electron gas, intensive investigations have been performed on small Al clusters9–17 and Au clusters (or on their charged, doped, or ligated counterparts),17–23 so as to highlight the fillings of electronic shells and hence to design new superatoms. A popular example is Al13 superatom in configuration of 1S21P61D102S21F142P5, which behaves like a halogen atom and exhibits a high electron affinity.11 However, due to the structural distortions stemmed from the Jahn–Teller effect,24 the filling of electronic orbitals in an ordinary superatom does not follow the Hund's rule of maximizing spin, and superatoms always favor nonmagnetic character with the paired electrons. These observations inspire an important issue whether nonmagnetic superatoms could be extended to magnetic species, with the intrinsic spin moments originating from superatom shells or with the introduced spin moments originating from localized atomic d states, i.e., how to tune the magnetic properties of superatoms? According to the employed method commonly for designing magnetic materials, single transition-metal atom (usually 3d or 4d TM atom) was extensively encapsulated into small simple-metal clusters, where host matrixes are composed of elements of group-IA (Li,25 Na,26–29 K,29 Cs27,29), group-IIA (Mg,30–33 Ca,34 Sr,35,36), group-IIIB (Al,11–13,17 Ga,17,37), and group-IVB (Si,38–40 Ge,40,41 Sn,40,41 Pb42,43). The prototype of magnetic superatom is V-alkali clusters (VLi8, VNa8 and VCs8) in configuration of 1S21P61D5, which can mimic a manganese atom and show a large spin magnetic moment of 5 μB.25–27 So far, it is generally accepted that in magnetic superatoms, atomic d-state electrons (or superatomic D states) localized on TM sites would provide spin magnetic moment, while the delocalized s, p-valence electrons of simple-metal atoms and of TM atom would fully occupy diffuse superatomic S, P states and provide superatomic stability. In addition, under the consideration that stable superatoms can compose superatomic molecules by sharing their valence pairs, alternative efforts have been devoted to superatomic molecules to explore the promising hints for designing novel superatom-assembling materials.22,32,44 Very recently, the newly constructed Li14, Li10, Li8 clusters are proposed to be analogues to conventional F2, N2, CH4 molecules owning to the similar exhibitions both in molecular-orbital diagrams and bonding-patterns.44
Following the original motivation for developing new superatoms, spherical core–shell structures by placing small cluster into large cage at high symmetry have been designed to rich superatom family, such as [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12− in bulk A12Cu12Sn21 intermetallics45–47 as well as A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd, Mn) in cluster phase.48 Unfortunately, their endohedral icosahedra commonly shares an empty or closed-shell d10 configuration, or d electrons are not taken into account for filling the superatomic orbitals. In this content, investigations should be done to explore whether above experience can be extended to other systems, in particular to a further suitable cluster/cage combination. What remained to be determined is which primary cage together with suitable encapsulation would be better for imparting novel superatoms. The dodecahedral Bi20-fullercage49 and icosahedral TM13 clusters50–56 have been previously determined to be very stable and low-lying energetically (not necessarily the lowest), and the isolate dodecahedral Bi20-fullercage is the most stable one among nitrogen family of N, P, As, Sb cages.49 Consequently, systemic encapsulations of TM13 into Bi20 would provide an opportunity to select a suitable elemental cluster in achieving the maximum strength of Bi–TM bond, because radii of core TM cluster is in progression from left to right for a row of the periodic table. Indeed, we have previously determined that for a given composition, the most favored structure of bimetallic BiMn and BiCo clusters tend to be an icosahedral TM13-core encapsulated into a dodecahedral Bi-cage.57 Such core–shell structure at high symmetry may offer the possibility of altering properties nearly at will by varying the number of itinerant electrons yet not breaking the electronic degeneracy.
With these views in mind, we perform a systematic density functional theory (DFT) investigations on the three-shell icosahedral matryoshka clusters [TM@TM12@Bi20]−, where TM atom encompasses every one of 3d and 4d periods. The key objective of current work is to explore the possibility of cluster being as superatom. What role, if any, does the electronic structure play? It further prompts the question, how general are the alignments of superatom orbitals in these clusters? For example, would they always hold true for different constituents regardless of the magnetic moment? The remaining of this paper is organized as follows: in Sec. II, we show the theoretical approach and computational details. The results are reported in Sec. III. Section IV contains a discussion and our conclusions.
2 Computational method
The representative structures of TM13 and [TM13@Bi20]− (TM = 3d, 4d) clusters are depicted in Fig. 1, where an icosahedral TM13 fragment resides at the center of Bi20 dodecahedral cage (fullerene) to give an onion-skin-like structure [TM@TM12@Bi20]−. Icosahedron TM12 and pentagonal dodecahedron Bi20 are reciprocal structures by switching their face-centers and vertexes,45,58 i.e., each TM atom of the intermediate TM12 shell is at the center of each Bi5 ring of the outer Bi20 shell and, in turn, each Bi atom is on the top of one TM3 triangular face. Ultimately, 32 surface atoms form a dimpled geodesic sphere in Ih symmetry and it is composed of 60 triangular faces. The geometrical optimizations are performed at the constrained Ih symmetry and the initially imposed high spin moments. In an effort to confirm the magnetic ground state, allowing spin multiplicities are further examined on the optimal structure by alternating the values above and below the obtained spin multiplicity.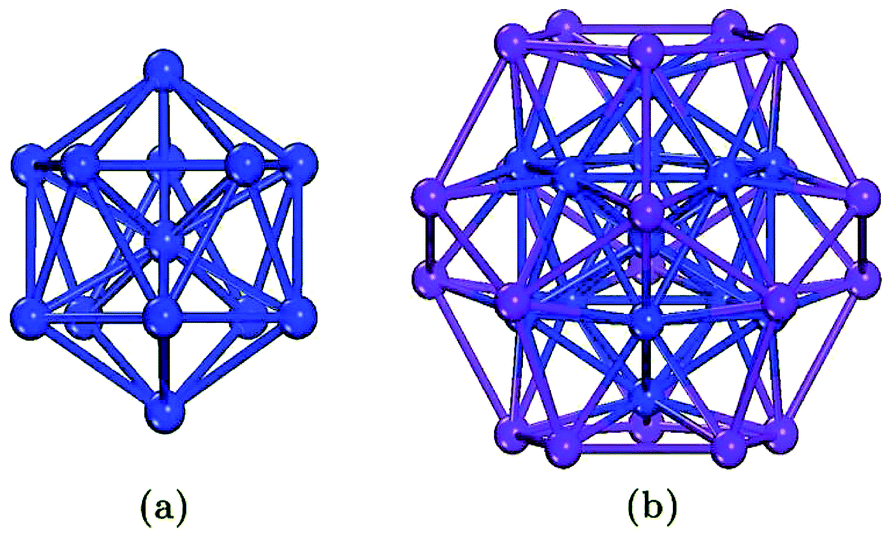 | ||
| Fig. 1 Representative structure of TM13− cluster (a) and [TM13@Bi20]− intermetalloid (b) in Ih symmetry. | ||
The DFT optimizations are carried out by using the DMOL simulation package,59 employing the effective core potential (ECP),60,61 double-numerical basis sets with p-polarization function (DNP), and PW91 exchange–correlation potential under the generalized-gradient-approximation (GGA). The 3d4s, 4d5s, 6s6p electrons are respectively treated as the valence for 3d and 4d TM atoms and Bi atom, and relativistic effect at the scalar relativistic level is taken into account in the calculations. The direct inversion in iterative sub-space (DIIS) approach together with the thermal smearing scheme with a value of 0.005 Ha is adopted to speed up the self-consistent filed (SCF) convergence. The convergence criteria of 1 × 10−5 Ha Å−1 for the energy gradient and 5 × 10−3 Å for the atomic displacement are forced in structural optimizations. The SCF charge density is converged to 1 × 10−6 e Å−1, which allows a total energy convergence of 1 × 10−5 Ha. The density of states (DOS) was obtained by a Gaussian extension of the discrete energy levels and summation over them. The broadening width parameter was chosen to be 0.2 eV. The calculations of the atomic contributions to molecular orbitals of Sc13@Bi20 cluster to support the inspection of MO plots have been also performed with Gaussian 09 software at GGA/BPW91/6-311+G* level,62 and molecular orbitals visualization are performed by using the Molekel software.63 The corresponding energy eigenvalues can be grouped into atomic-like shells (1S, 1P, 1D, 2S, 1F, 2P, …), depending on the degeneracy and spatial symmetry of the molecular orbitals. The lower case letters (s, p, d, …) are used to denote electronic levels in atoms and upper case letters (S, P, D, …) to denote the superatomic shells of electrons in a confined nearly-free electron gas.
3 Results and discussion
3.1 Stability analysis
For the endohedral [TM13@Bi20]− clusters, we show in Table 1 the distances from central TM atom to intermediate TM atom (Rcore) and to outer Bi atom (Rshell), binding energy (Eb), embedding energy (De), charge transfer (Q), HOMO–LUMO gap (gap), and electron affinity (EA). Corresponding values of anionic TM13− clusters are also presented as a reference. Thermal stability is estimated in terms of the binding energy Eb = (12ETM + ETM−1 − Ecluster)/13 for anionic TM13− cluster and Eb = (12ETM + ETM−1 + 20EBi − Ecluster)/33 for anionic [TM13@Bi20]− cluster, where ETM, ETM−1, and EBi are the energies of neutral TM, anionic TM, and neutral Bi atoms, respectively, and Ecluster is total energy of cluster. The embedding energy (De), defined as De = Eshell + Ecore − Eendo, represents the gain in energy as TM13− cluster is encapsulated into Bi20 cage, where Eshell, Ecore, Eendo denote the optimal energies of Bi20 cage, TM13−1 icosahedra, and endohedral [TM13@Bi20]− system, respectively. For empty Ih-Bi20 cage, our optimizations show that it has Bi–Bi bond-length of 3.04 Å and binding energy of 2.16 eV per atom, which agrees well with previous DFT values on the same structure.49 For icosahedral TM13− clusters, the overall trends of binding energy as the function of element are consistent with the trends of previous works.50–52,56| TM13− | [TM13@Bi20]− | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TM | R | Eb | Gap | Rcore | Rshell | L | Eb | De | QBi | QTM | QTM′ | Gap | EA | M |
| Sc | 3.03 | 2.80 | 0.32 | 3.09 | 4.94 | 3.10 | 3.20 | 26.02 | −0.29 | 0.42 | −0.25 | 0.21 | 3.33 | 0 |
| Ti | 2.63 | 3.67 | 0.97 | 2.76 | 4.70 | 3.01 | 3.66 | 29.80 | −0.18 | 0.20 | 0.04 | 0.48 | 2.86 | 3 |
| V | 2.43 | 3.34 | 0.27 | 2.61 | 4.60 | 2.98 | 3.48 | 28.45 | −0.13 | 0.16 | −0.33 | 0.23 | 2.55 | 14 |
| Cr | 2.71 | 1.70 | 0.14 | 2.79 | 4.64 | 2.96 | 2.51 | 17.44 | −0.17 | 0.21 | −0.11 | 0.14 | 2.84 | 13 |
| Mn | 2.64 | 1.84 | 0.12 | 2.71 | 4.60 | 2.95 | 2.87 | 27.47 | −0.11 | 0.11 | −0.13 | 0.37 | 3.42 | 36 |
| Fe | 2.39 | 3.58 | 0.49 | 2.56 | 4.52 | 2.93 | 3.18 | 15.34 | −0.13 | 0.03 | 1.32 | 0.25 | 2.73 | 33 |
| Co | 2.36 | 3.70 | 0.40 | 2.44 | 4.48 | 2.95 | 3.20 | 14.19 | −0.07 | 0.04 | −0.08 | 0.17 | 2.04 | 18 |
| Ni | 2.36 | 3.03 | 0.25 | 2.53 | 4.45 | 2.89 | 3.30 | 26.53 | −0.13 | 0.13 | 0.07 | 0.12 | 2.85 | 5 |
| Cu | 2.46 | 2.15 | 0.73 | 2.55 | 4.52 | 2.94 | 2.49 | 10.93 | −0.03 | 0.03 | −0.09 | 0.98 | 1.71 | 4 |
| Zn | 3.01 | 0.66 | 1.99 | 2.78 | 4.73 | 3.03 | 1.50 | −2.27 | −0.10 | 0.10 | −0.13 | 0.10 | 2.42 | 0 |
| Y | 3.33 | 3.04 | 0.04 | 3.42 | 5.19 | 3.23 | 3.32 | 26.82 | −0.52 | 0.80 | −0.19 | 0.21 | 3.92 | 0 |
| Zr | 2.91 | 4.66 | 1.28 | 3.07 | 4.90 | 3.12 | 4.09 | 31.30 | −0.30 | 0.42 | −0.02 | 0.64 | 3.02 | 3 |
| Nb | 2.77 | 4.65 | 0.38 | 2.93 | 4.76 | 3.01 | 4.02 | 29.12 | −0.30 | 0.41 | 0.17 | 0.25 | 2.77 | 0 |
| Mo | 2.59 | 3.50 | 0.13 | 2.78 | 4.64 | 2.96 | 3.56 | 28.95 | −0.21 | 0.24 | 0.33 | 0.12 | 2.51 | 7 |
| Tc | 2.52 | 4.26 | 0.26 | 2.68 | 4.58 | 2.94 | 3.89 | 29.66 | −0.04 | −0.04 | 0.17 | 0.16 | 2.30 | 4 |
| Ru | 2.54 | 4.28 | 0.14 | 2.77 | 4.53 | 2.87 | 4.19 | 39.38 | 0.04 | −0.15 | −0.01 | 0.17 | 2.18 | 11 |
| Rh | 2.58 | 3.79 | 0.11 | 2.90 | 4.54 | 2.85 | 3.02 | 7.11 | 0.05 | −0.18 | 0.07 | 0.11 | 2.62 | 8 |
| Pd | 2.68 | 2.40 | 1.45 | 3.00 | 4.59 | 2.86 | 2.87 | 20.22 | 0.06 | −0.18 | −0.04 | 0.42 | 2.55 | 0 |
| Ag | 2.83 | 1.60 | 0.75 | 2.92 | 4.74 | 3.00 | 2.37 | 14.25 | 0.00 | −0.06 | −0.26 | 0.76 | 1.96 | 6 |
| Cd | 3.38 | 0.59 | 2.13 | 3.13 | 4.99 | 3.14 | 1.19 | −11.64 | −0.11 | 0.11 | −0.23 | 0.16 | 2.58 | 5 |
In Fig. 2a and b, TM13− and [TM13@Bi20]− clusters are compared to each other in Eb values as a function of constituent element. The analogous oscillation suggests that thermal stability of [TM13@Bi20]− is primarily determined by TM13 fragment but weakly effected by outer Bi20 cage. On the one hand, two humps appear at Ti, Fe, Co, and Ni sites on Eb curve of [TM13@Bi20]− clusters with TM = 3d series (Zr, Nb, and Ru sites for TM = 4d series), indicating that these doping counterparts are more thermally stable than others. Although there are a few relatively stable cases, most [TM13@Bi20]− have the Eb over 3.0 eV (Table 1) that are generally large than the reported values of superatomic A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd, Mn).48 From a comparison of Eb values of [TM13@Bi20]− between 3d and 4d counterparts given in Table 1 and shown in Fig. 2, the 3d doping ones often give smaller value than the 4d doping ones do for the early-half elements (i.e., from Sc to Fe for 3d series vs. from Y to Ru for 4d series), while it is substantially inverse for the late-half elements (i.e., from Co to Zn for 3d series vs. from Rh to Cd for 4d series). Even for the relatively large values at 3d elements of Ti (3.66 eV), Fe (3.18 eV), Co (3.20 eV), Ni (3.30 eV), they are still smaller than the relatively small values at 4d elements of Mo (3.56 eV) and Tc (3.89 eV). On the other hand, the positive De verifies the feasibilities that most TM13− species can be attractively embedded into Bi20 cage (except for the negative values at Zn and Cd), and the large values at Ti, Fe, Mn, Ni for 3d series (Zr, Nb, and Ru for 4d series) reinforce the reliable assignment that they are more suitable in doping into Bi20 cage to form stable [TM13@Bi20]−.
 | ||
| Fig. 2 The binding energy (a and b) and HOMO–LUMO gap (c and d) as a function of TM element. | ||
The outer- and inner-cages with optimal matching size are crucial for designing a stable onion-skin-like structure, and thus it is indispensable to analyze their cage-radii. Between outer-Bi20-shell and intermediate-TM12-icosahedra, the bond-lengths of Bi–TM range from 2.85 Å to 3.23 Å, which are somewhat longer than the typical values of bimetallic dimers. In fact, strong interactions between TM and Bi atoms can be discerned by the contractive distance between outer-shell and intermediate-shell: before doping, Router between centroid-site and surface Bi is 4.26 Å for Bi20 cage (Rcore ranges from 2.36 Å to 3.38 Å for TM13−); after doping, Router and Rcore are respectively elongated to 4.45–5.19 Å and 2.44–3.42 Å for endohedral [TM13@Bi20]−. By calculating the elongated differences in radii (Δ2R = ΔRshell − ΔRcore), one can estimate the magnitude of their interactions because small Δ2R is related to short TM–Bi bond-length. The variations of Δ2R along the elemental atoms are plotted in Fig. 3, where small values are at middle period. This can be related to that more d electrons take part in chemical bonds to strengthen TM–Bi bond-length. In addition, Δ2R present a similar trend for 3d and 4d period owing to an analogous evolution of electronic configurations.
 | ||
| Fig. 3 The difference of the elongated radii Δ2R = ΔRshell − ΔRcore. | ||
The chemical stabilities of [TM13@Bi20]− are examined via the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The HOMO–LUMO gaps for 3d (4d) doping series are plotted in Fig. 2c (2d), where the values at Ti, Mn, and Cu for 3d elements (Zr and Ag for 4d elements) are remarkably larger than that at others. The superior chemical stability of these systems can be related to the contributions of electronic structure since their structural configurations are same. Interestingly, TM13− and [TM13@Bi20]− generally exhibit a comparable value of HOMO–LUMO gap, indicating the prominent role of TM13− in determinations of electronic structure. For TM = nd10 elements, TM13− are found to be an insulator (gaps are 1.99 eV and 2.13 eV for Zn13− and Cd13−) while [TM13@Bi20]− counterparts are almost a metal (corresponding values decrease to 0.10 eV and 0.16 eV). The is attributed to the hybridization effect between sp states of TM atoms and 6p states of Bi atoms, e.g., the HOMO-level of Cd13− cluster (5s states) is lower in energy than the HOMO-level of Bi20 cage (6p state), but an opposite case is found for the LUMO-level (5p state at high level for Cd13− vs. 6p states at low level for Bi20). Once Cd13− is doped into Bi20 cage, 6p electrons from HOMO state of Bi20 would form a new occupied-orbital (being as the HOMO of [TM13@Bi20]−) and LUMO state of Bi20 would form a new unoccupied-orbital (being as the LUMO of [TM13@Bi20]−), resulting in the decreased gap from 2.13 eV for Cd13− to 0.16 eV for [Cd13@Bi20]−. The HOMO and LUMO of [Cd13@Bi20]− are mostly contributed from Bi-6p states can be evidenced by their electron density, i.e., it is found that the accumulated charges are mainly around the Bi atom in [TM13@Bi20]−. Further verifying of gaps via the partial DOS will be discussed in the following section, where the HOMO and LUMO of [Sc13@Bi20]− mainly consist of 6p orbitals of Bi atoms.
The moderate gaps 0.37–0.98 eV are presented by [TM13@Bi20]− (TM = Ti, Mn, Cu, Zr, Ag) clusters, which are much smaller than the values of previously proposed nonmagnetic superatoms, i.e., [As@Ni12@As20]−3 (1.46 eV),45,46 [Sn@Cu12@Sn20]−12 (1.34 eV),47 and A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd) (1.27–1.54 eV).48 One thing should be emphasized here that, atoms within empty or closed-shell d10 configuration have been merely considered in previous works (d electrons are not taken into account for filling the superatomic orbitals). Since the energy gaps between adjacent energy levels decrease continuously with the increase of quantum numbers, it is no surprise that [TM13@Bi20]− systems having more valence electrons (fill to the superatomic orbitals with high quantum numbers) would present somewhat small HOMO–LUMO gaps with respect to that of the systems having less valence electrons (fill to the superatomic orbitals with low quantum numbers). In addition, there is a strong tendency that the standard GGA and LDA approximations to DFT often underestimate the gaps while hybrid functionals such as PBE0,64,65 HSE06,66 and B3LYP47 will predict slightly more accurately. This may also be the reason we obtained smaller gaps at standard GGA level. Nevertheless, if the energy gaps of the previously proposed magnetic superatoms are taken as the references, our moderate values of specific magnetic clusters are always comparable to their reports, i.e., V@Na8 (0.42 eV),26 Sc@K12 and Sc@Cs12 (0.35 eV),29 Fe@Ca8 (0.61 eV),30 Fe@Mg8 (0.64 eV),32 Tc@Mg8 (0.65 eV),33 Mn@Ca9 (0.92 eV),34 Mn@Sr9 (0.39 eV)36 as well as the analogous structures of [Pb@Mn12@Pb20] (0.61 eV) and [Sn@Mn12@Sn20] (0.38 eV).48
3.2 Electron filling rule
For an ideal spherical structure like a free atom, its highly degenerate atomic orbitals would resemble the spherical harmonics and could be identified with spherical electronic shells (1S21P61D102S21F142P61G182D103S21H222F143P61I242G18…, or alternation of the sequence of electronic shells); however, for a nearly spherical structure like an icosahedron, the degeneracy in some electronic states will be broken by the rearrangement of internal ionic forming synthetic cluster. Owning to icosahedral ligand field of [TM13@Bi20]− structure (the highest symmetry axis is in 5-fold), the molecular orbitals within a high angular momentum that is more than 5-fold degenerate (labeled by F, G, H…) will split into a set of sub-orbitals. According to the irreducible representations of point group Ih symmetry, generally speaking, S-type orbitals show ag symmetry, P-type orbitals show as t1u symmetry, D-type orbitals transform as hg, F-type orbitals split into two sets transforming as gu + t2u or gu + t1u, G-type orbitals split into two sets of gg + hg symmetry, and H-type orbitals split into three sets of t2u + gg + gu or t2u + t1u + hu symmetry.47In an effort to evaluate the superatomic characteristics of a series of [TM13@Bi20]− clusters, [Sc13@Bi20]− cluster is firstly exemplified to illustrate the filling rule of valence electrons into each molecular orbital in the consideration of its total 140 valence electrons. Free Bi and Sc atoms are in the configuration 6s26p3 and 3d14s2, and Bi20 and Sc13 fragments would provide 100 and 39 valence electrons to valence pool, respectively. Charge density isosurfaces and one-electron energy levels for the Kohn–Sham molecular orbitals of [Sc13@Bi20]− cluster are presented in Fig. 4 and 5, respectively, where each molecular orbital is distinguishingly assigned by the inspection of their nodes, shapes, and energetic sequence analogous to that of atomic orbitals. From Fig. 4, one can note that the spatial shapes of each molecular orbital resemble the corresponding atomic orbital, e.g., the lowest energy state is spread out over the entire cluster accommodating 2 electrons, and thus it can be regarded as the 1S superorbital. Except for the low-lying states from 1S to 1F that are fairly diffused over Bi outer-shell and can be deemed to be the contributions of Bi-6s electrons, other states have the large amplitudes on both Bi and TM sites, indicating the strong hybridizations between Bi-6p states and TM-3d states. In addition, the molecular orbitals within high angular momentum come to split into a set of suborbitals as analyzed above. In brief, the filling of 70 superatomic orbitals is proposed to in energy sequence of 1S2(ag)1P6(t1u)1D10(hg)1F8(gu)1G8(gg) 1F6(t2u)2S2(ag)1G10(hg)2P6(t1u)1H6(t2u)1H10(hu)2G8(gg)3S2(ag)2D10(hg)2F8(gu)2H6(t2u)2H8(gg)2F6(t1u)2H8(gu)2G10(hg). Nevertheless, it has been proposed that D supershell is followed by F supershell in a homogeneous confined nearly free electron gas (jellium model for a homogeneous cluster); it however leads to 2G over 2D and 2F set of orbitals in bimetallic heterogeneous clusters.
 | ||
| Fig. 4 The molecular orbital charge density isosurfaces (iso = 0.001 a.u. for 1S, 2S and 3S orbitals; 0.002 a.u. for 1P, 1D, 1F, 2P, 2D, 2F and 3D orbitals; 0.02 a.u. for the 1H, 2H, 1G and 2G orbitals) for [Sc13@Bi20]−. | ||
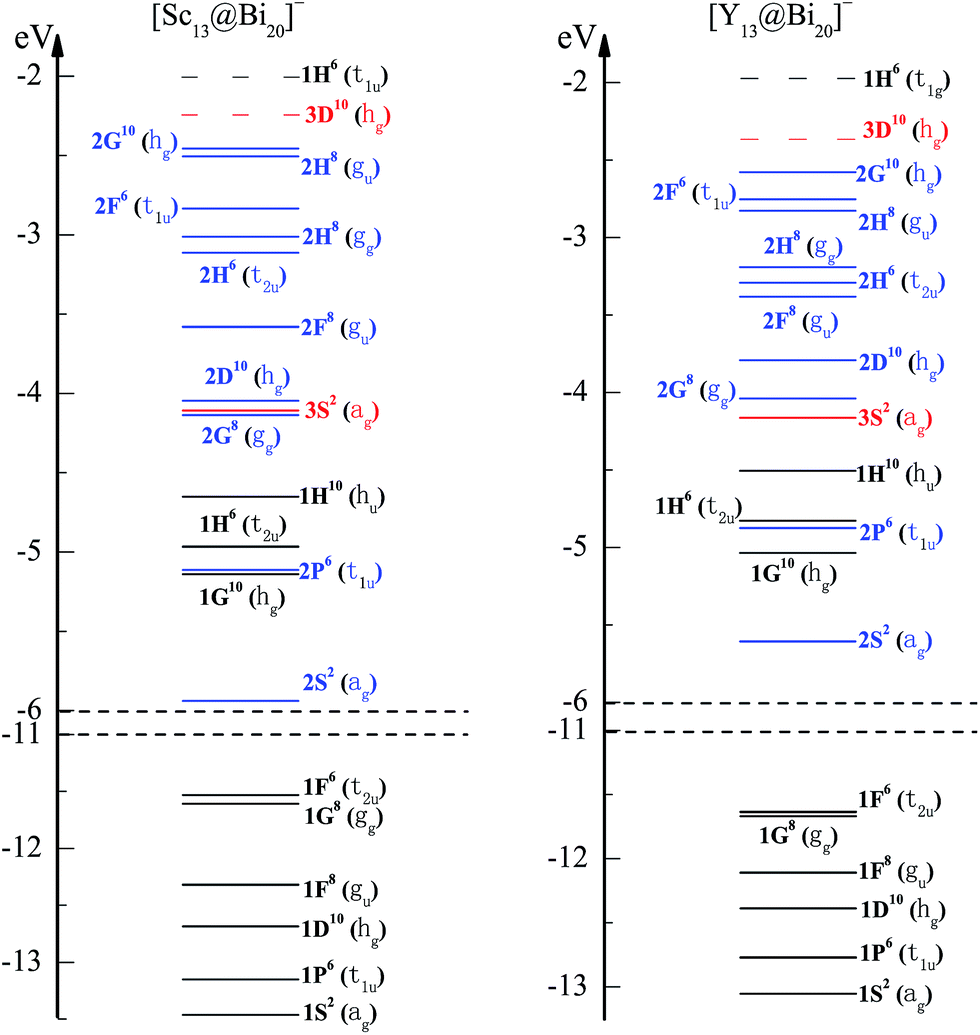 | ||
| Fig. 5 The energy levels (in eV) of molecular orbitals of [Sc13@Bi20]− and [Y13@Bi20]−, which are assigned to a series of superatomic orbitals corresponding to the indicated spherical harmonics. Continuous lines correspond to the occupied levels, whereas the broken lines correspond to the unoccupied states. For each level, the angular character (with upper-case letter), total electrons (with superscript), and degenerated states (within parenthesis) are marked. | ||
In Fig. 5, the first shell n = 1 of the occupation 1S21P61D101F141G181H16 shows an incompletely filled 1H orbitals (fully filled 1H sub-shell needs 22 electrons): 1H molecular orbitals split into one 5-fold degenerate sub-orbitals and two 3-fold degenerate sub-orbitals; the 5-fold degenerate sub-orbitals 1H10(hu) and the low-lying 3-fold degenerate sub-orbitals 1H6(t2u) are fully occupied by 16 electrons while the high-lying 3-fold degenerate sub-orbitals 1H6(t2u) are above the Fermi level and empty. Except for 1G8(gg) and 1F6(t2u) orbitals that are almost equal in energy, the orbital sets of Bi-6s block (1S⋯1F orbitals) are energetically well separated from each other. The second shell n = 2 of the occupation 2S22P62D102F142G182H22 as well as the third shell n = 3 of the occupation 3S2, show both closure sub-shell configurations. There is a modest energy separation of 0.21 eV between the HOMO of 2G10(hg) and LUMO of 3D10(hg). This filling configuration is reminiscent of atomic situation in Mg atom ([Ne]3s2). By comparing the electronic shells of [Y13@Bi20]− with its iso(valence) electronic counterpart of [Sc13@Bi20]−, one found that the molecular orbitals of [Y13@Bi20]− are arranged in analogy to those of [Sc13@Bi20]− with two exceptions that the energy orders of 2G8(gg) ⇌ 3S2(ag) orbitals as well as 2F6(t1u) ⇌ 2H8(gu) orbitals are alternated. Generally speaking, quantum states of these two clusters can be bunched into supershells with associated orbitals having the shapes resembling those in atoms, and they can be classified as superatoms.
Fig. 6 shows the partial density of states (PDOS) projected at Sc and Bi sites of [Sc13@Bi20]−. Below the energy level −11.0 eV, 20 lowest-lying orbitals of 1S21P61D101F141G8 are primarily accommodated by 2 × 20 = 40 6s valence electrons from Bi20 skeleton frame. This is consistent with previous findings that for a polyhedral cluster with structure vertices occupied by “p-block” element atoms, their lowest-lying “S-block” molecular orbitals mainly comprise atomic s orbitals of “p-block” element atoms.47,67,68 In the combination of Fig. 5 and 6, it is clear that the following orbitals are dominated by the contributions of Bi-6p, Sc-4s, Sc-3d electrons (3 × 20 + 3 × 13 + 1 = 100): 2S2 orbital at −6.0 eV is filled by Sc-4s states with Bi-6p characters admixing (2 electrons); 1G102P61H6 orbitals around −5.0 eV and 2G83S22D10 orbitals around −4.0 eV have contributions from Bi-6p states mixing with Sc-4s and Sc-3d states (42 electrons); 2F8 orbitals at −3.5 eV are merely composed of Bi-6p states (8 electrons); the remaining orbitals 1H102H82H62F62H82G10 (48 electrons) are basically from Bi-6p and Sc-3d states in strong hybridizations. On the whole, PDOS ranging from −6.0 to −2.0 eV show a high share of Bi-6p contributions, and the occupied electrons may be described as the Bi–Bi surface bonding in main.
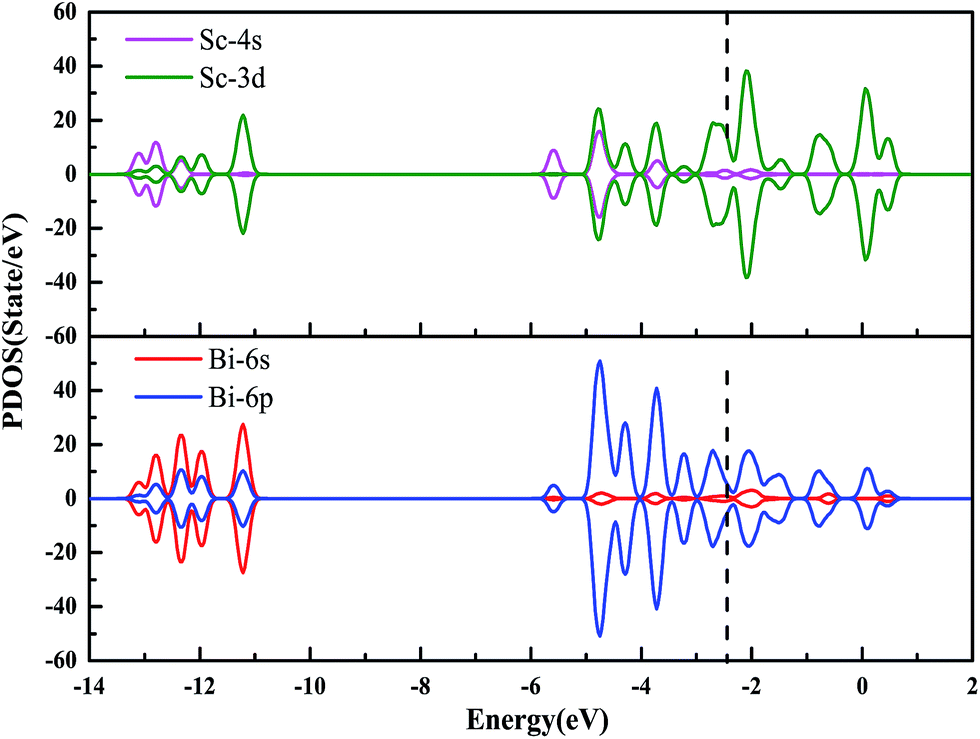 | ||
| Fig. 6 Spin-polarized partial density of states (PDOS) for [Sc13@Bi20]−. The HOMO level is marked by the dotted line. | ||
Previously, electronic account scheme on [As@Ni12@As20]3− has been proposed by Moses and Zhao et al.,45,46 i.e., 60 of 100 valence electrons of As20 pentagonal dodecahedron are involved in 30 As–As bonds along 30 edges, and the remaining 40 are attributed to 20 As-pz orbitals oriented radially with respect to cluster surface and termed as ąřpz lone-pair electronsąś. They have also determined somewhat radial interactions between 20 As-pz orbitals of As20 and virtual orbitals of As@Ni12, together with the negligible interactions between 30 As–As bonding orbitals of As20 and Ni–Ni bonding orbitals of As@Ni12. In terms of above electronic account scheme, on the one hand, if 40 6s-electrons of outer Bi20 are regarded as pz-like lone-pair electrons (60 6p-electrons are as Bi–Bi bonding electrons), the 6s states of outer Bi20 cage should interact with s and d states of inner Sc13 fragment. However, from the PDOS of [Sc13@Bi20]− in Fig. 6, no hybridization between Bi-6s and Sc-3d, 4s is discerned. On the other hand, if 40 6p-electrons of outer Bi20 are regarded as the pz lone-pair electrons, then the residual 20 6p-electrons together with 40 electrons from Bi-6s states are involved in forming Bi–Bi bonds. Unexpectedly, 40 6s-electrons do not participate in Bi–Bi or Bi–Sc bonds from the PDOS. As a matter of fact, our previous DFT calculations on Bi clusters69 have revealed that the average Mayer bond-orders between Bi atoms decrease from 3 in Bi2 dimer to 1 in Bi4 tetrahedral and to 0.89 in Bi24 chain-like cluster (average coordination number increases from 1 to 3 and to 3.05), suggesting that three 6p electrons of each Bi atom are merely being ready to form chemical bonds while two 6s electrons are being as lone pairs. In addition, DFT investigations on BiMn clusters also determined no hybridization between Bi-6s states and Mn-3d, 4s, 4p states.57 Therefore, above electronic account scheme is not applicable for [Sc13@Bi20]− cluster and for Bi-based relevant [TM13@Bi20]− clusters.
In addition, considerable elongations of Bi–Bi bond-length in [TM13@Bi20]− clusters require less 6p-electrons to form weak Bi–Bi bonds, and more remaining 6p-electrons would exist as the lone pairs in pz orbitals (i.e., less than 60 6p-electrons are required to form 30 dodecahedral Bi–Bi edges, and then more remaining 6p-electrons are as lone-pair electrons to form Bi–TM bonds.) Our calculations on the Mayer bond-orders of [TM13@Bi20]− clusters show that the bond-orders between the nearest neighbor Bi atoms range from 0.32 to 0.38, indicating that nearly one electrons of each Bi atom would take part in the formation of three Bi–Bi bonds. Consequently, 20 6p-electrons of Bi20 are indeed as Bi–Bi bonding electrons and 40 6p-electrons are as pz lone-pair electrons to form Bi–TM bonds. This partitioning scheme of Bi20 electrons is in agreement with the population analyses. According to on-site population analysis (by Mulliken definition), TM-atoms of intermediate shell often partially donate their s electrons to Bi-atom 6p and 6d orbitals, while most centered TM-atom d and p orbitals gains electrons. The situations come to reverse for Ru, Rh, Pd, Ag dopants, where Bi-atoms donate their electrons to outer-TM12 and inner-TM atoms (Table 1). The charge transferring indicates the occurrence of hybridizations between Bi-p, TM-s, p, and d states, and thus all clusters share the same pattern of chemical bonding. However, no matter what kind of situations, the numbers of Bi-atom 6s electrons always maintain the same, suggesting no interactions between Bi-atom 6s state and other states.
3.3 Magnetic analysis
Superatomic cluster is remarkably stable because of its filling electronic shells with paired electrons, corresponding to a nonmagnetic state such as nonmagnetic [Sc13@Bi20]− cluster. Here, we will focus on Mn13 endohedral [Mn13@Bi20]− cluster, based on the magnetic fact that [Mn13@Au20]−, [Pb@Mn12@Pb20], and [Sn@Mn12@Sn20] were predicted to possess giant magnetic moments of 44, 28, and 28 μB,48,70 respectively. Previously, magnetic superatoms can be achieved by doping a singly magnetic TM atom into a small simple-metal cluster, because either superatomic D-state has strong component from atomic d-state (i.e., d electrons are regarded as the delocalized valences in filling superatomic shells) or superatomic D-state hybridize strongly with d-states (i.e., d electrons are regarded as the localized valences and do not fill superatomic shells).26–36 Above both cases can result in the exchange-splitting between the majority and minority states either of superatomic D shells or of atomic d shells, leading to a superatom with a spin magnetic moment. Unfortunately, the maximum magnetic moments in these systems are often less than 6 μB, since the numbers of their valence electrons are not enough to fill superatomic shells large than nD (the largest spin moment is 5 μB when the degenerated 1D orbital is half-filled). Even if some systems can provide enough valence electrons to fill superatomic shells with large orbital quantum numbers, owning to the potential field splitting of ionic cores of their oblate shape, they would break the degeneracy of orbitals into several groups of sub-orbitals. This would lower the prospective magnetic moments further. One may question whether or not above deficiency could be overcome by a large cluster with an utmost spherical configuration. Consequently, iso-structures of [Mn13@Bi20]− is quite intriguing to represent a distinct magnetic moment among 3d and 4d doping series (Table 1). It should be mentioned again that the geometrical optimizations are first done by ferromagnetically imposing maximum spin moment on each TM atom in terms of Hund's first rule so as to search for the structural minima, and then the energy minimizations for a number of fixed total spin moments are orderly executed by alternating the spin moments above and below the obtained value in first step. If the total energy decreases with the variations of total spin moments of cluster, further spin state will be considered until the energy minimum with respect to ground spin state is reached. In addition, for clusters having sizeable spin values in first optimization step, various spin arrangements are also tried in a quest for the spin ground state.Fig. 7 presents the PDOS and spin density distribution, which is associated with each orbital level for [Mn@Mn12@Bi20]−. As demonstrated by the spin density distribution in Fig. 7, the magnetism mainly localizes on Mn atoms. By analyzing the PDOS in addition, three interesting things can be discerned: (1) the alignment of superatomic orbitals of magnetic [Mn13@Bi20]− still resembles those of non-magnetic [Sc13@Bi20]−, supporting the universal filling principle of valence electrons into superatomic shell for different [TM13@Bi20]−; (2) 3F, 3G, 3H, 4S, 4P, 4D superatomic states are dominantly from the contributions of Mn-3d states and Bi-6p states; (3) large exchange-splitting between the majority and minority superatomic states happens to occur, coincidentally with large exchange-splitting of Mn-3d states. For example, the majority 3F⋯4D sub-shells are fully filled while the corresponding minority counterparts are fully empty. In addition, Mulliken population analysis shows again that 3F⋯4D states have large contribution from the Mn-d states, i.e., magnetic moment is mainly localized on Mn atom (3.82 μB per Mn atom of intermediate Mn shell; −4.22 μB of central Mn), and a small amount of antiferromagnetic moment is found on outer Bi atoms (−0.28 μB per Bi atom). Therefore, one can reasonably deduce that the splitting in Mn-3d state is the driving force for the splitting of supershells. The full-filled shells within 156 electrons (1S21P61D101F141G181H16‖2S22P62D102F142G182H22‖3S23P63D10), together with the half-filled majority orbitals within 36 alpha electrons (3F7α3G9α3H11α‖4S1α4P3α4D5α) and the full-empty corresponding minority orbitals, lead to [Mn13@Bi20]− a stable species having a moderate HOMO–LUMO gap of 0.37 eV. The majority 3F⋯4D superatomic states of [Mn13@Bi20]− give a giant total magnetic moment of 7 + 9 + 11 + 1 + 3 + 5 = 36 μB. This exemplifying study on magnetic [Mn13@Bi20]− reinforces our finding that the filling of superatomic shells is also appreciate for magnetic [TM13@Bi20]− species, and it can be classified as magnetic superatoms.
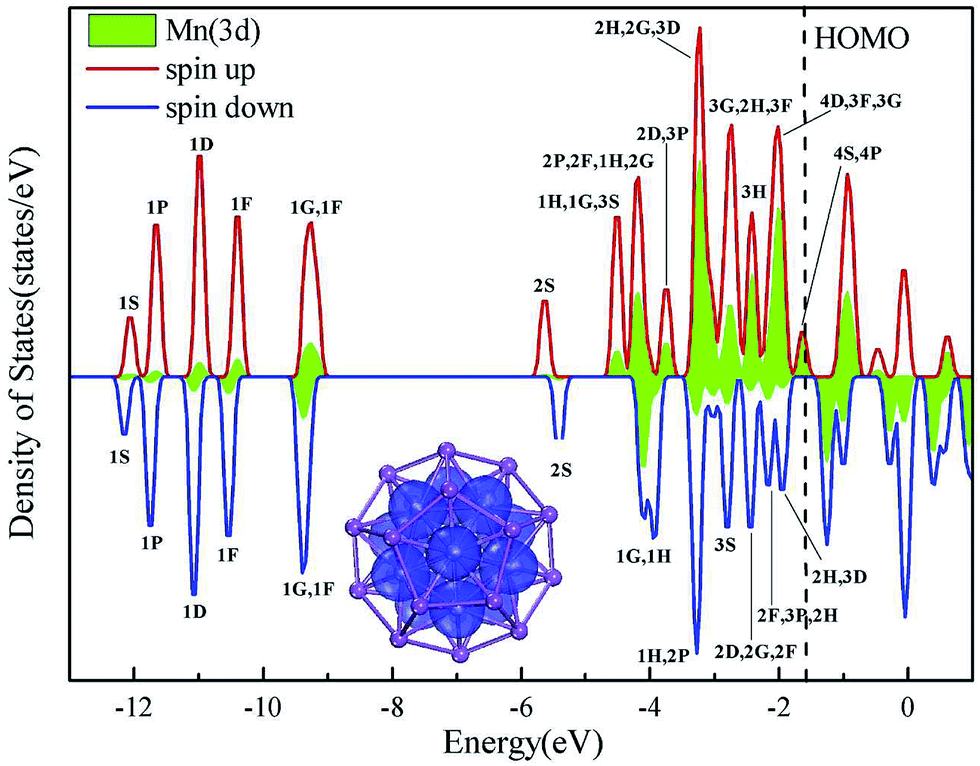 | ||
| Fig. 7 The occupied superatom orbitals are labeled on the spin-polarized density of states (DOS) for [Mn13@Bi20]−, where the d states from Mn atoms are filled by green color. The isosurface of spin density distribution is presented (blue areas) as insert chart. | ||
The [Fe13@Bi20]− has 13 more addition valence electrons compared to [Mn13@Bi20]−, for which 5 of 13 electrons would like to fill into one of the splitted majority sub-orbitals 4Gα5↑ (4Gα9 → 4Gα5 + 4Gα4) while the remaining 8 electrons would like to fill into the minority orbitals 4Sβ1 4Pβ3 3Gβ4. This results in a closed-shell configuration within 164 valence electrons (1S21P61D101F141G181H16‖2S22P62D102F142G182H22‖3S23P63D104S24P6) and the opened-shell configuration having 37 alpha and 4 beta electrons (3Fα73Gα93Hα11‖4Dα54Gα53Gβ4). Consequently, [Fe13@Bi20]− has total valence electrons of 205 and magnetic moment of 33 μB. For [Co13@Bi20]− within additional 26 electrons compared to [Mn13@Bi20]− (218 total valence electrons), 4 electrons fill into the majority subshell 4Gα4 to form a configuration of 3Fα73Gα93Hα11‖4Sα14Pα34Dα54Gα4, and 22 valence electrons fill into the minority subshell 3Fβ73Gβ53Hβ6‖4Sβ14Pβ3 (3Gβ9 → 3Gβ5 + 3Gβ4; 3Hβ11 → 3Hβ3 + 3Hβ3 + 3Hβ5), resulting in a magnetic moment of 18 μB. Nevertheless, the partially filled sub-shells lead to somewhat small gaps of 0.25 eV and 0.17 eV for [Fe13@Bi20]− and [Co13@Bi20]−, respectively. From the Table 1, as we go across the 3d period series after Mn element, more additional electrons would like to occupy the minority orbitals than the majority orbitals, giving rise to decreasing numbers of unpaired electrons and thus the decreasing magnetic moments along elemental table. Strangely, although Cu13, Ag13 and Cd13 are nonmagnetic species, [TM13@Bi20]− can present modest magnetic moments of 4 μB, 6 μB and 5 μB at Cu, Ag and Cd cases, respectively. This means that even if there is no driving force from magnetic TM13 to induce magnetism on [TM13@Bi20]−, superatoms can also yield magnetic moment through the exchange splitting between the majority and minority states of superatomic orbitals. Therefore, this work offers two strategies to design magnetic superatom by the improvement of exchange splitting between majority and minority superatomic states: one is by magnetic TM13 fragment inducement; another one is originated from the partial fillings of superatomic shells to result in unpaired electrons following Hund's rule.
4 Conclusion
To develop the novel superatoms, a systemic study of structural, electronic, and magnetic properties of icosahedra TM13− (TM = 3d, 4d) clusters encapsulated into pentagonal dodecahedra Bi20 cage has been performed by using the DFT method. Unlike the cases in previous works, here, we design a framework by invoking systems that take d electronic states as valence electrons to fill the superatomic orbital states. The stability of [TM13@Bi20]− cluster is analyzed in terms of binding energy, embedding energy, and HOMO–LUMO gap, and the results show that most TM13− species can be attractively embedded into Bi20 cage to form a stable three-shell icosahedral matryoshka cluster. Following the definition of superatom of jellium model, we propose that [TM13@Bi20]− clusters can mimic the atomic patterns in electronic shells in both molecular diagrams and energy consequences, and they share an analogy behavior of superatomic orbitals. It is determined that the partial d electrons bring the magnetic moments by improving the exchange-splitting of superatomic orbitals of high angular momentum, while the remaining d electrons together with s, p electrons contribute to the stability by filling the superatomic orbitals to form the full-filled or half-filled subshells. Stable [Mn13@Bi20]− cluster is predicted to exhibit large magnetic moment (36 μB) and moderate HOMO–LUMO gap owning to its half-filled superatomic subshell, and thus it is being as a protyle magnetic superatom.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project No. 10904125 and 91121013), the National College Students' Innovation and Entrepreneurship Training Program (Project No. 20130635060), and the Fundamental Research Funds for the Central Universities (Project No. XDJK2012B008).References
- M. Brack, Rev. Mod. Phys., 1993, 65, 677–732 CrossRef CAS.
- W. A. de Heer, Rev. Mod. Phys., 1993, 65, 611–676 CrossRef CAS.
- W. D. Knight, K. Clemenger, W. A. de Heer Saunders, M. Y. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141–2143 CrossRef CAS.
- S. N. Khanna and P. Jena, Phys. Rev. Lett., 1992, 69, 1664–1667 CrossRef CAS PubMed.
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS.
- A. W. Castleman Jr and S. N. Khanna, J. Phys. Chem. C, 2009, 113, 2664–2675 CrossRef.
- A. W. Castleman Jr, S. N. Khanna, A. Sen, A. C. Reber, M. Qian, K. M. Davis, S. J. Peppernick, A. Ugrinov and M. D. Merritt, Nano Lett., 2007, 7, 2734–2741 CrossRef PubMed.
- C. Ashman, S. N. Khanna, F. Liu, P. Jena, T. Kaplan and M. Mostoller, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 15868–15875 CrossRef CAS.
- C. Y. Cha, G. Ganteför and W. Eberhardt, J. Chem. Phys., 1994, 100, 995–1010 CrossRef CAS.
- R. E. Leuchtner, A. C. Harms and A. W. Castleman Jr, J. Chem. Phys., 1989, 91(4), 2753–2754 CrossRef CAS.
- D. E. Bergeron, A. W. Castleman Jr, T. Morisato and S. N. Khanna, Science, 2004, 304, 84–87 CrossRef CAS PubMed.
- X. Li, H. Wu, X. B. Wang and L. S. Wang, Phys. Rev. Lett., 1998, 81, 1909 CrossRef CAS.
- D. E. Bergeron, P. J. Roach, A. W. Castleman Jr, N. O. Jones and S. N. Khanna, Science, 2005, 307, 231–235 CrossRef CAS PubMed.
- J. U. Reveles, S. N. Khanna, P. J. Roach and A. W. Castleman Jr, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18405–18410 CrossRef CAS PubMed.
- M. Akutsu, K. Koyasu, J. Atobe, N. Hosoya, K. Miyajima, M. Mitsui and A. Nakajima, J. Phys. Chem. A, 2006, 110, 12073–12076 CrossRef CAS PubMed.
- P. A. Clayborne, O. Lopez-Acevedo, R. L. Whetten, H. Grönbeck and H. Häkkinen, J. Chem. Phys., 2011, 135, 094701 CrossRef PubMed.
- O. Lopez-Acevedo, P. A. Clayborne and H. Häkkinen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 035434 CrossRef.
- P. Pyykkö and N. Runeberg, Angew. Chem., 2002, 114, 2278–2280 CrossRef.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 CrossRef CAS PubMed.
- J. Nishigaki, K. Koyasu and T. Tsukuda, Chem. Rec., 2014, 14, 897–909 CrossRef CAS PubMed.
- M. Walter and H. Häkkinen, Phys. Chem. Chem. Phys., 2006, 8, 5407–5411 RSC.
- S. N. Khanna, B. K. Rao, P. Jena and J. L. Martins, Physics and Chemistry of Small Clusters, ed. P. Jena, B. K. Rao and S. N Khanna, Plenum Press, New York, 1987, pp. 435–438 Search PubMed.
- M. Zhang, J. Zhang, X. Feng, H. Zhang, L. Zhao, Y. Luo and W. Cao, J. Phys. Chem. A, 2013, 117, 13025–13036 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, H. Wang, A. Lim, G. Gantefoer, K. H. Bowen, J. U. Reveles and S. N. Khanna, J. Am. Chem. Soc., 2013, 135, 4856–4861 CrossRef CAS PubMed.
- J. U. Reveles, P. A. Clayborne, A. C. Reber, S. N. Khanna, K. Pradhan, P. Sen and M. R. Pederson, Nat. Chem., 2009, 1, 310–315 CrossRef CAS PubMed.
- J. U. Reveles, P. Sen, K. Pradhan, D. R. Roy and S. N. Khanna, J. Phys. Chem. C, 2010, 114, 10739–10744 CrossRef CAS.
- K. Pradhan, J. U. Reveles, P. Sen and S. N. Khanna, J. Chem. Phys., 2010, 132, 124302 CrossRef PubMed.
- V. Chauhan, V. M. Medel, J. U. Reveles, S. N. Khanna and P. Sen, Chem. Phys. Lett., 2012, 528, 39 CrossRef CAS.
- V. M. Medel, A. C. Reber, J. U. Reveles and S. N. Khanna, J. Chem. Phys., 2012, 136, 134311 CrossRef PubMed.
- V. M. Medel, J. U. Reveles, S. N. Khanna, V. Chauhan, P. Sen and A. W. Castleman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10062–10066 CrossRef CAS PubMed.
- G. X. Ge, Y. Han, J. G. Wan, J. J. Zhao and G. H. Wang, J. Chem. Phys., 2013, 139, 174309 CrossRef PubMed.
- V. M. Medel, J. U. Reveles, M. F. Islam and S. N. Khanna, J. Phys. Chem. A, 2013, 117, 4297–4303 CrossRef CAS PubMed.
- V. Chauhan and P. Sen, Chem. Phys., 2013, 417, 37–44 CrossRef CAS.
- V. M. Medel, J. U. Reveles and S. N. Khanna, J. Appl. Phys., 2012, 112, 064313 CrossRef.
- J. Hartig, A. Stösser, P. Hauser and H. Schnöckel, Angew. Chem., Int. Ed., 2007, 46, 1658–1662 CrossRef CAS PubMed.
- H. Cantera-López, L. C. Balbás and G. Borstel, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 075434 CrossRef.
- A. K. Singh, V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115429 CrossRef.
- V. Kumar and Y. Kawazoe, Appl. Phys. Lett., 2003, 83, 2677–2679 CrossRef CAS.
- V. Kumar and Y. Kawazoe, Appl. Phys. Lett., 2002, 80, 859–861 CrossRef CAS.
- S. Neukermans, E. Janssens, Z. F. Chen, R. E. Silverans, P. V. R. Schleyer and P. Lievens, Phys. Rev. Lett., 2004, 92, 163401 CrossRef CAS PubMed.
- B. Zhou, T. Krämer, A. L. Thompson, J. E. McGrady and J. M. Goicoechea, Inorg. Chem., 2011, 50, 8028–8037 CrossRef CAS PubMed.
- L. Cheng and J. Yang, J. Chem. Phys., 2013, 138, 141101 CrossRef PubMed.
- M. J. Moses, J. C. Fettinger and B. W. Eichhorn, Science, 2003, 300, 778–780 CrossRef CAS PubMed.
- R. B. King and J. Zhao, Chem. Commun., 2006, 4204–4205 RSC.
- S. Stegmaier and T. F. Fässler, J. Am. Chem. Soc., 2011, 133, 19758–19768 CrossRef CAS PubMed.
- X. Huang, J. Zhao, Y. Su, Z. Chen and R. B. King, Sci. Rep., 2014, 4, 6915 CrossRef CAS PubMed.
- A. D. Zdetsis, J. Phys. Chem. C, 2010, 114, 10775–10781 CrossRef CAS.
- Y. Sun, M. Zhang and R. Fournier, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075435 CrossRef.
- Y. Sun, R. Fournier and M. Zhang, Phys. Rev. A, 2009, 79, 043202 CrossRef.
- M. Zhang and R. Fournier, Phys. Rev. A, 2009, 79, 043203 CrossRef.
- H. K. Yuan, H. Chen, A. L. Kuang, C. L. Tian and J. Z. Wang, J. Chem. Phys., 2013, 139, 034314 CrossRef CAS PubMed.
- H. K. Yuan, H. Chen, A. L. Kuang, A. S. Ahmed and Z. H. Xiong, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 174412 CrossRef.
- H. K. Yuan, H. Chen, A. S. Ahmed and J. F. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 144434 CrossRef.
- M. J. Piotrowski, P. Piquini and J. L. F. Da Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155446 CrossRef.
- H. Chen, H. K. Yuan, A. L. Kuang, Y. Miao, P. Chen and Z. H. Xiong, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 184429 CrossRef.
- G. Bergman, J. L. Waugh and L. Pauling, Acta Crystallogr., 1957, 10, 254–259 CrossRef CAS.
- DMOL, V960, Biosym Technologies, San Diego, CA, 1996 Search PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- M. J. Frisch, Gaussian 09, revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- U. Varetto, Molekel 5.4.0.8, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- P. Liu, J. Nisar, R. Ahuja and B. Pathak, J. Phys. Chem. C, 2013, 117, 5043–5050 CrossRef CAS.
- P. Liu, J. Nisar, B. Pathak and R. Ahujaab, Phys. Chem. Chem. Phys., 2013, 15, 17150–17157 RSC.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fäsler, Angew. Chem., Int. Ed., 2011, 123, 3712–3754 CrossRef.
- B. Wahl, M. Erbe, A. Gerisch, L. Kloo and M. Ruck, Z. Anorg. Allg. Chem., 2009, 635, 743–752 CrossRef CAS.
- H. K. Yuan, H. Chen, A. L. Kuang, Y. Miao and Z. H. Xiong, J. Chem. Phys., 2008, 128, 094305 CrossRef CAS PubMed.
- J. Wang, J. Bai, J. Jellinek and X. C. Zeng, J. Am. Chem. Soc., 2007, 129, 4110–4111 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |