
Photocatalytic direct intermolecular addition of 1,3-dithianes†
Ming Lee,
Ying-Ho Chen,
Tzu-Hang Hung,
Wen Chang,
Wei-Cheng Yan and
Dasheng Leow*
Department of Chemistry, National Tsing Hua University, 101, Sec 2, Kuang-Fu Rd., Hsinchu 30013, Taiwan. E-mail: dsleow@mx.nthu.edu.tw
First published on 8th October 2015
Abstract
A direct intermolecular addition of 1,3-dithianes to alkenes has been accomplished via visible-light mediated photocatalysis. This new process proceeds in moderate to excellent yields and eliminates the need for pre-functionalization of substrates. Electronic tuning of the 1,3-dithiane plays a vital role in ensuring reactivity. Both aromatic and aliphatic aldehyde-derived 1,3-dithianes are compatible. This radical umpolung strategy allows the synthesis of γ-keto-α-amino acid derivatives.
Umpolung or polarity inversion is a powerful strategy in turning an acyl group into a nucleophile by converting it into a 1,3-dithiane. Corey and Seebach are the first to recognize the potential usefulness of this concept.1 Subsequently Smith's group developed the dithianes as linchpins.2 On the other hand, the electrophilicity of 1,3-dithianes has been relatively less explored. Nakayama and co-workers first prepared 1,3-dithiolyium salts and reported their electrophilic reactions.3 Stahl et al. also observed similar reactivity in these compounds and reported their results over the years.4
The last mode of reactivity of 1,3-dithianes is the radical pathway, and it is also the least known among the three modes in the literature. There has been scattered reports in the past 20 years and the majority of them required the pre-installation of a radical initiator group at the 2-position to generate the C-2 centered radicals. Several radical initiator groups such as phenylseleno,5a xanthates,5b TEMPO,5c and chloro5d,e were known. The need for radical initiator groups sometimes severely limits the substrate scope and synthetic applications.
 | (1) |
 | (2) |
 | (3) |
 | (4) |
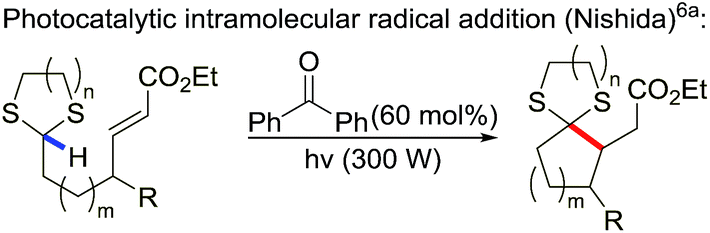
There are precedent examples of direct radical addition of 1,3-dithianes to alkenes but they are restricted to intramolecular events. Nishida and co-workers reported an elegant work using benzophenone as photocatalyst for the 1,3-dithiane intramolecular direct addition to alkene functional group (eqn (1)).6a Curran's group also demonstrated that this type of reaction could proceed using tin hydride/AIBN method.6b Roberts et al. found that tri-tert-butoxysilanethiol and peroxyketal radical initiator could catalyze the radical cyclization of aldehydes and acetal via the in situ formation of 1,3-dithiane.6c,d To the best of our knowledge, the direct intermolecular radical addition of 1,3-dithiane to alkene remains elusive and challenging.
We were inspired by Ishii's work on N-hydroxyphthalimide (NHPI)-catalyzed hydroxyacylation of alkenes using 1,3-dioxolanes under aerobic conditions (eqn (2)).7,8 They demonstrated that heteroatoms stabilized radicals generated from the homolytic cleavage of C–H were able to participate in intermolecular addition to alkenes. However the main drawback of this reaction is that huge excess of substrate has to be used as solvent. This will be problematic when the starting material is in limited quantity.
In fact, many alternative solutions such as carbene catalysis,9 rhodium catalysis,10 and acyl radical chemistry11 have been developed for hydroacylation of aldehydes to alkenes. Photocatalytic methods also exist for hydroacylation reaction of alkenes. Fagnoni and co-workers reported this reaction using decatungstate photocatalyst,12a while Maruoka's group employed hypervalent iodine(III) catalyst.12b However these methods are ineffective for aromatic aldehydes. On the other hand, Fu et al. achieved the decarboxylative 1,4-addition of α-oxocarboxylic acids but their method is restricted to aromatic substituents.12c
Visible light photoredox catalysis is gaining chemists' attention due to its various advantages such as mild conditions as well as high selectivities.13,14 On the basis of these prior observations, we hypothesize that visible-light photocatalysis may be able to provide a solution to the challenging problem of direct intermolecular radical addition of 1,3-dithiane to alkenes. As part of our ongoing photocatalysis program, herein we disclose our findings.15
We started off our studies using dithiane 2m and methyl acrylate 3a as the model reaction (Scheme 1). Dithiane 2m was adapted from the original Corey–Seebach reaction. Unfortunately all our attempts to make it react in the presence of various photocatalysts such as 1g proved to be futile. Therefore we turned to the literature and found that in the Kellogg's reaction, 2,3-dihydrobenzothiazoles were oxidized by activated halides catalyzed by photocatalyst Ru(bpy)3Cl2 1a (eqn (3)).16 We decided to tune the electronic properties by switching to 1,3-benzodithioles, which was more electron-withdrawing (eqn (4)). This protecting group was first introduced by Pelter's and Smith's groups and subsequently utilized by others.17
 | ||
| Scheme 1 Failed attempt using Corey–Seebach dithiane 2m. | ||
Initially, the model reaction between dithiane 2a and alkene 3a failed to react with several photocatalysts (Table 1, entries 2–4). To our delight, photocatalyst 1f [E1/2(M*/M−) = 1.21 V vs. SCE]13b gave respectable yield. The yield was further increased by changing the counteranion (Table 1, entry 7–8). These results showed that the large redox potential of photocatalyst 1g was crucial (see Table S1†). Switching from CFL to LEDs, which were UV/IR-free, resulted in similar yields (Table 1, entry 9). Reaction did not proceed in the absence of light at 40 °C (Table 1, entry 10). Adding extra bases did not lead to an increase in yield (Table S3†).
|
|||||
|---|---|---|---|---|---|
| Entry | Photocat. (mol%) | Yield (%) | Entry | Photocat. (mol%) | Yield (%) |
| a Unless otherwise noted, the reaction conditions were as followed: dithiane 2a (0.10 mmol), methyl acrylate 3a (0.5 mmol), photocatalyst (1 mol%), anhydrous DMF (1.0 mL), 24 h, ambient temperature, irradiated with 23 W 6500K compact fluorescent light (CFL) under N2 atmosphere.b Yield determined by 1H NMR analysis of unpurified reaction mixture using MeNO2 as internal standard.c 13 W LEDs was used instead of 23 W CFL.d Reaction conducted in dark at 40 °C. | |||||
| 1 | None | <5 | 6 | 1e | <5 |
| 2 | 1a Ru(bpy)3Cl2 | <5 | 7 | 1f | 68 |
| 3 | 1b rose bengal | 7 | 8 | 1g | 75 |
| 4 | 1c eosin Y | <5 | 9c | 1g | 71 |
| 5 | 1d lr(ppy)3 | <5 | 10d | 1g | <5 |

With optimized conditions in hand, we went on to examine the scope of this reaction (Table 2). The dithianes bearing aromatic substituents generally gave moderate to good yields (4a–f). This reaction worked even better with alkyl substituents (4g–l). Various functional groups such amide and ether groups were tolerated (4i, j). The reaction even proceeded with a bulky group (4l).


Next we went on to examine this reaction with a variety of olefins (Table 3). Other electron-withdrawing groups such as cyano or ketone worked excellently (4m, n). Various 1,1-disubstituted alkenes also proceeded smoothly (4o–t). The reaction was highly accommodative towards hydrogen donors like acetamide group (4s). Due to the nucleophilic nature of the radicals,6a electronically unbiased olefins (styrenes) failed to react. This reaction was also sensitive to steric hindrance at the reacting site of alkene.


Natural amino acids bearing an oxo group at γ position such as L-kynurenine play important roles in various biological functions. Hence we investigated the synthetic applications of the alkylated products. We scaled up the reaction of dithiane 2h with alkene 3r for the synthesis of amino acid derivative 4r (Scheme 2). During this scale-up, the catalytic loading was lowered to 0.5 mol% yet the reaction yield was maintained at 92%. We managed to isolate 0.5 grams of the dithiane 4r. Following the deprotection of dithiane 4r, γ-keto-α-amino acid derivative 5 was achieved in 80% yield.
 | ||
| Scheme 2 Practical synthesis of γ-keto-α-amino acid derivative. | ||
For preliminary mechanistic studies, we found that the reaction was completely inhibited by TEMPO free radical (1 equiv.). Stern–Volmer fluorescence quenching experiments confirmed that the emission of excited state of Ir(III) was quenched by dithiane 2c, which followed a linear concentration-dependent correlation. We also conducted the light/dark experiment and confirmed that visible light was necessary for the reaction to progress.
Based on our preliminary experimental and previous observations,6,14f a plausible reaction pathway was proposed (Scheme 3). In the first step, the excited state of photocatalyst was quenched by dithiane 2 and radical intermediate 6 was formed by single electron transfer and deprotonation.18 Radical 6 was trapped alkene 3 to form radical 7 intermediate. Then it was reduced by the photocatalyst and re-protonation gave the final alkylated product 4 along with the regeneration of photocatalyst 1g.
 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In conclusions, we have developed a visible-light mediated photocatalyzed direct intermolecular radical addition of 1,3-dithiane to alkenes under ambient temperature, low catalytic loading, and neutral conditions. This reaction is applicable for a variety of aldehyde-derived 1,3-dithianes, including aromatic and aliphatic examples. Another feature of this reaction is that it is compatible with many functional groups. This is highlighted by the synthesis of α-amino acids containing an oxo group at the γ position. The underlying key factor to successful reaction is the use of the unique 1,3-benzodithioles, which is modified from the original Corey–Seebach dithiane.Acknowledgements
We gratefully acknowledge National Tsing Hua University (NTHU) and the Ministry of Science and Technology of Taiwan (102-2113-M-007-017-MY2) for financial support.Notes and references
- For reviews, see: (a) B. T. Gröbel and D. Seebach, Synthesis, 1977, 6, 357 CrossRef; (b) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef PubMed.
- For reviews, see: (a) A. B. Smith III, S. M. Condon and J. A. McCauley, Acc. Chem. Res., 1998, 31, 35 CrossRef; (b) A. B. Smith III and C. M. Adams, Acc. Chem. Res., 2004, 37, 365 CrossRef PubMed.
- (a) J. Nakayama, K. Fujiwara and M. Hoshino, Chem. Lett., 1975, 1099 CrossRef CAS; (b) J. Nakayama, K. Fujiwara and M. Hoshino, Bull. Chem. Soc. Jpn., 1976, 49, 3567 CrossRef CAS.
- For selected papers, see: (a) I. Stahl and I. Kühn, Chem. Ber., 1983, 116, 1739 CrossRef CAS PubMed; (b) I. Stahl, Chem. Ber., 1985, 118, 1798 CrossRef CAS PubMed; (c) I. Stahl, Chem. Ber., 1985, 118, 3159 CrossRef CAS PubMed.
- (a) J. H. Byers, C. C. Whitehead and M. E. Duff, Tetrahedron Lett., 1996, 37, 2743 CrossRef CAS; (b) M. de Greef and S. Z. Zard, Tetrahedron, 2004, 60, 7781 CrossRef CAS PubMed; (c) A. J. Herrera and A. Studer, Synthesis, 2005, 1389 CAS; (d) W. Du, L. Tian, J. Lai, X. Huo, X. Xie, X. She and S. Tang, Org. Lett., 2014, 16, 2470 CrossRef CAS PubMed; (e) W. Du, J. Lai, L. Tian, X. Xie, X. She and S. Tang, Chem. Commun., 2014, 50, 14017 RSC.
- (a) A. Nishida, M. Nishida and O. Yonemitsu, Tetrahedron Lett., 1990, 31, 7035 CrossRef CAS; (b) D. P. Curran and W. Shen, J. Am. Chem. Soc., 1993, 115, 6051 CrossRef CAS; (c) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC; (d) H.-S. Dang and B. P. Roberts, Tetrahedron Lett., 1999, 40, 8929 CrossRef CAS.
- For a review, see: Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393 CrossRef CAS.
- (a) K. Hirano, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2000, 2457 RSC; (b) P. Du, H. Li, Y. Wang, J. Cheng and X. Wan, Org. Lett., 2014, 16, 6350 CrossRef CAS PubMed.
- For selected examples, see: (a) M. S. Kerr, J. R. de Alaniz and T. Rovis, J. Am. Chem. Soc., 2002, 124, 10298 CrossRef CAS PubMed; (b) K. Hirano, A. T. Biju, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 14190 CrossRef CAS PubMed.
- For a review, see: (a) M. C. Willis, Chem. Rev., 2010, 110, 725 CrossRef CAS PubMed. For selected examples, see: (b) C.-H. Jun, H. Lee and J.-B. Hong, J. Org. Chem., 1997, 62, 1200 CrossRef CAS; (c) A. H. Roy, C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 2082 CrossRef CAS PubMed; (d) M. M. Coulter, K. G. M. Kou, B. Galligan and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16330 CrossRef CAS PubMed.
- For a review, see: (a) D. L. Boger and R. J. Mathvink, J. Org. Chem., 1992, 57, 1429 CrossRef CAS; For selected examples, see: (b) S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352 RSC; (c) W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756 CrossRef CAS PubMed.
- (a) S. Esposti, D. Dondi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 2531 CrossRef CAS PubMed; (b) S. A. Moteki, A. Usui, S. Selvakumar, T. Zhang and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 11060 CrossRef CAS PubMed; (c) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Svoboda and B. König, Chem. Rev., 2006, 106, 5413 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- For selected examples, see: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed; (c) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed; (d) H. Liu, W. Feng, C. W. Kee, Y. Zhao, D. Leow, Y. Pan and C.-H. Tan, Green Chem., 2010, 12, 953 RSC; (e) D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed; (f) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed; (g) X. Liu, X. Ye, F. Bureš, H. Liu and Z. Jiang, Angew. Chem., Int. Ed., 2015, 54, 11443 CrossRef CAS PubMed.
- (a) D. Leow, Y.-H. Chen, T.-H. Hung, Y. Su and Y.-Z. Lin, Eur. J. Org. Chem., 2014, 7347 CrossRef CAS PubMed; (b) D. Leow, Org. Lett., 2014, 16, 5812 CrossRef CAS PubMed; (c) Y.-H. Chen, M. Lee, Y.-Z. Lin and D. Leow, Chem.–Asian J., 2015, 10, 1618 CrossRef CAS PubMed.
- S. H. Mashraqui and R. M. Kellogg, Tetrahedron Lett., 1985, 26, 1453 CrossRef CAS.
- (a) S. Ncube, A. Pelter, K. Smith, P. Blatcher and S. Warren, Tetrahedron Lett., 1978, 19, 2345 CrossRef; (b) D. Petruzziello, A. Gualandi, H. Giaffar, V. Lopez-Carrillo and P. G. Cozzi, Eur. J. Org. Chem., 2013, 4909 CrossRef CAS PubMed.
- For a review on thiyl radicals, see: F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data for all new compounds. See DOI: 10.1039/c5ra19069j |
| This journal is © The Royal Society of Chemistry 2015 |