DOI:
10.1039/C5RA19015K
(Paper)
RSC Adv., 2015,
5, 105400-105408
De novo design of stereochemically-bent sixteen-residue β-hairpin as a hydrolase mimic†
Received
15th September 2015
, Accepted 1st December 2015
First published on 3rd December 2015
Abstract
De novo design is a powerful tool to explore principles of protein folding and function. Design of enzymes is a formidable challenge: a designed polypeptide must fold to bind the desired guest, catalyze the desired reaction, release the product formed, and accomplish all the effects simultaneously. Here we describe design of a stereochemically-bent sixteen-residue β-hairpin polypeptide as a hydrolase mimic. The enzyme is accomplished to the desired specificity in two steps; first main chain is organized as the desired fold, and then sequences are optimized to the desired specificity as enzymes. The fold is designed stereochemically and the enzyme is achieved chemically: stereochemical modification of a canonical poly-L β-hairpin furnishes the desired fold which is optimized to the desired sequence specificity as an enzyme. The optimization of sequence implemented by inverse design is targeted for having the desired fold programmed as the desired enzyme chemically. Distinct side chains defining the desired active site are reshuffled to have their roles in stabilizing the targeted enzyme and in specifying it as a catalyst tested. The designed sequences of heterochiral structure are synthesized and are assessed for substrate binding by fluorescence and for enzymatic activity with UV-based kinetic assays. We report success in achieving hydrolases in sixteen-residue polypeptides with application of p-nitrophenyl acetate as the model substrate. The enzyme mimics, even while modest in activity, are remarkable in their short sequence length relative to natural enzymes, which are constrained to be long polypeptides and homochirality of poly-L structures could be a reason. The study thus underscores the value stereochemistry offers for having novel folds fashioned, and the value stepwise algorithm offers to have the folds of desired function specificity programmed chemically.
Introduction
De novo design is motivated primarily to test the understanding of protein folding, stability, and function. The approach targets complimentary objectives: design of known or novel structure, and test of the folding and functional principles. The design of proteins of arbitrary three-dimensional structure remains a grand challenge. The problem is simplified by its reduction into two steps: selection of tertiary structure and design of a compatible amino acid sequence. Remarkable progress has been made in mastering the second step. Successful methods have been developed for computing new sequences for the tertiary structures of interest.1–4 Notable examples were the pioneering redesign of a zinc finger protein by Mayo,1 the redesign of function by alteration of binding specificity by Hellinga,5 and the design of enzyme-like proteins of altered specificity by Bolon and Mayo.6 The methods, however, do not create new protein structures from scratch; rather, the natural scaffolds are remodeled to the desired specificity chemically, either in the core for alteration of protein stability or in active site for reengineering of function.6–11 The ability to alter protein stability or function is valuable, but the scope remains limited to the natural protein scaffolds. Design of the folds not encountered in Nature remains a challenge.
The natural building block monomers of proteins remain the L-α-amino acids. The D-enantiomers have been explored and can, as inserts into poly-L sequences, diversify shapes of the molecular folds stereochemically.12 The diversity allowed by stereochemistry as the design variable may enhance design specifically in accomplishing greatly simplified structures to a diversity of molecular shapes. Illustrating the concept, canoe13 of 20 residues was designed as a mimic of the protein cationophore “catgrip”, and π-cup14 of 15 residues was designed as a synthetic receptor for acetylcholine as the binding ligand. The design of canonical β-hairpin was reported as a remarkably aggregation-free protein effective as a hydrolase against an acetylcholine surrogate.15 The design of zinc-finger hydrolase was reported as an αββ fold that tri-coordinates zinc to activate its function as an enzyme.16
Enzyme design is a formidable challenge; sequences must be chosen to fold predictably, to bind the chosen substrate correctly, to catalyze the desired reaction specifically, and to release the product for regeneration of the enzyme.17–21 Significant progress in addressing the multifactorial challenge has been made in recent years.22–34 In the present study, we approach the design in steps, in first fold and then sequence. For fold design stereochemistry is applied as the variable, and for sequence design amino acid side chains are applied as the variable. Poly-L sequences of a canonical β-hairpin are thus reprogrammed first as the desired morphological folds stereochemically, and then as the desired enzymes chemically. The polypeptides thus accomplished by stepwise design are proven to manifest the desired function specificity as enzymes. The enzymes are modest in catalytic power, but are remarkably short polypeptides relative to the natural poly-L congeners, which typically are long polypeptides and for the reasons that remain largely obscure. The study affirms stereochemistry as the variable to approach the folds of greatly diversified morphological possibility and greatly minimized molecular structure as proteins. The study also illustrates the algorithm to have the protein of simplified and greatly articulated structure to be optimized as the desired structure and functions specificity chemically. Given unusual size and simplicity of the enzyme mimics accomplished, the study furnishes promising candidates for possible future evolution to enhance catalysis power and possible analysis of the basis with rigor.
Results
Design and synthesis
The design was approached in a two-step algorithm: first a molecular fold was targeted stereochemically; next a sequence was targeted for the desired specificity of enzyme catalysis chemically. The β-hairpin fold targeted for design as a protein was nucleated over a DPro8-LGly9 dipeptide unit; thus the structure was promoted to ordering as a type II′ β-hairpin stereochemically.35 The flat ribbon-like structure of the propagating β-hairpin was mutated in a pair of cross-strand related L-residues to D-structure; the introduced ∼90° bend at the sequence positions 3 and 14 in the sixteen-residue hairpin created a local bend in the naturally flat structure. The aromatic residues Tyr, Phe, and Trp are positioned in concavity of the bent hairpin; thus possibility of π–π interactions is invoked for conferring conformational stability in the fold of interest. Besides locking the fold, the interaction of aromatic side chains are also deployed for binding a desired aromatic substrate on basis of π–π interactions. Enzyme catalytic groups in form of acid–base side chains of Asp or Glu and His, and hydroxyl side chain of Ser are placed suitably for the programmed fold to serve as a hydrolase for an ester group in the aromatic substrate.
The designed sequence plans are given in Table 1 and a cartoon representation of one of the designed polypeptides A1 is presented in Fig. 1. In A1, the planar ribbon of poly-L β-hairpin structure is modified with a local ∼90° bend with its cross-strand pair of LL-residues having been modified as the DD-structure. The bend, by placing aromatic and acid–base-nucleophile side chains in a desired spatial arrangement, may serve as the desired catalyst to hydrolyse an ester group in the active-site-anchored aromatic substrate. In A2, Glu replaces Asp in position 7 as an alternative acid group known to serve in the catalytic triad of human acetylcholinesterase enzyme.36 In A3, DLeu and DVal in sequence positions 3 and 14 are substituted with glycine to assess if the enhanced flexibility may impact catalysis function of the designed enzyme. In A4, the aromatic side chains for substrate binding and the cluster of functional side chains for catalyzed hydrolysis of the substrate are interchanged in the mutual positions. In A5, DPro in sequence position 8 is replaced with DAsn for assessing if the consequent labilization of β-hairpin as a conformational fold may impact its function as an enzyme.
Table 1 Sequences of designed sixteen-residue mixed chirality peptides
| Sequences |
| A1 |
Ac–Tyr1-Glu2-DLeu3-Asn4-Phe5-Ser6-Asp7-DPro8-Gly9-His10-Thr11-Ser12-Ala13-DVal14-Lys15-Trp16–NH2 |
| A2 |
Ac–Tyr1-Glu2-DLeu3-Asn4-Phe5-Ser6-Glu7-DPro8-Gly9-His10-Thr11-Ser12-Ala13-DVal14-Lys15-Trp16–NH2 |
| A3 |
Ac–Tyr1-Glu2-Gly3-Asn4-Phe5-Ser6-Asp7-DPro8-Gly9-His10-Thr11-Ser12-Ala13-Gly14-Lys15-Trp16–NH2 |
| A4 |
Ac–His1-Glu2-DLeu3-Asn4-Ser5-Ser6-Phe7-DPro8-Gly9-Trp10-Thr11-Tyr12-Ala13-DVal14-Lys15-Asp16–NH2 |
| A5 |
Ac–Tyr1-Glu2-DLeu3-Asn4-Phe5-Ser6-Asp7-DAsn8-Gly9-His10-Thr11-Ser12-Ala13-DVal14-Lys15-Trp16–NH2 |
 |
| | Fig. 1 Cartoon representation of computationally designed stereochemically-bent sixteen-residue β-hairpin peptide A1 as a hydrolase mimic. The cross-strand residues (Leu3 and Val14) mutated from LL-to-DD structure to promote a monomolecular fold, aromatic residues (Tyr1, Phe5 and Trp16) for substrate binding and catalytic residues (Asp7, His10 and Ser12) for substrate hydrolysis are shown in stick representation. | |
The peptides were made by manual solid-phase synthesis using standard Fmoc chemistry. In mass spectrum, ion peaks in correspondence of the expected molecular mass appeared in each peptide (Fig. S1†). 1H NMR spectra were recorded in 90% H2O/10% D2O at 298 K. The dispersal of chemical shifts in NMR spectrum of the structures implies that the synthesized peptides may be well ordered as specific folds (Fig. S2†). The sharpness of the resonances in the NMR spectrum implies also that the peptides may be freely soluble molecules. The spectra recorded at 2.5 mM and 0.25 mM concentration are free of any noticeable dilution-dependent variation, which affirms the absence of aggregation of the synthesized peptides in the concentration regime of NMR experiment.
Characterization of conformation
The conformation of peptides was assessed with CD. The spectra are characterized by appearance of minima of ellipticity in 200–205 nm range, except for A3 in which case a minimum appears at 197 nm (Fig. 2). While characteristic of β-sheet, the minima are blue shifted relative to the β-sheet minima in proteins, which typically appear at ∼217 nm. It has been recognized that the CD minimum in proteins tends to become blue shifted as the number of strands in the β-sheet construct are diminished. Thus the relatively blue shifted CD minimum of the peptides in this study is conformed to the presence of small number of β-strands in the structures. The polypeptides display an exciton couplet in 220–230 nm range (Fig. 2), implying that the aromatic groups are in mutual interaction.37,38 Mutual interaction among the groups that are terminal in the sequences implies that the designed sequences may be ordered as the planned β-hairpin folds. All peptides were practically concentration invariant in molar ellipticities in CD spectra (Fig. S3†); each folded molecule may be a monomer aggregation free in the concentration regime of CD experiment. The canonical β-hairpins of this length are notorious for aggregation due to β-sheet hydrogen bonds lateral to the hairpins and due to the side chains interacting facially between the flat hairpins.39–41 Such interactions of the canonical poly-L β-hairpins are thought to mediate their aggregation as amyloid plaques during the onset of neurodegenerative diseases.42–44 The lack of aggregation tendency in our synthetic β-hairpins, possibly due to their stereochemically bent structures, may reflect the break in continuous registry of hydrogen bonds and mutual stacking of side chains required for promotion of self-assembly.
 |
| | Fig. 2 CD spectra of peptides in water at 298 K. Y-axis represents the mean residue molar ellipticity and X-axis represents wavelength in nm. The peptide concentration is 40 μM. | |
Molecular dynamics studies for computation of conformational equilibria
In order to evaluate if the designed peptides do order like proteins, the structures were targeted for molecular dynamics (MD) simulations. The peptides were modeled as the stereochemically-bent β-hairpins and were in this conformation submitted to MD. The simulations were run for specific durations. Analysis of the trajectories (Table 2) implies that the designed sequences are appreciable in the potential for ordering as the desired folds (Fig. 3). The folds populating MD trajectories are locked with an average of 6.6–7.5 hydrogen bonds joining the peptides (Table 3). The hydrogen bonds locking the main chain are predominantly long ranged, as required for the β-hairpin, and about a fifth are medium ranged, as required for the β-turn.
Table 2 Statistics of conformers and microstates in molecular dynamics simulation of peptide variants
| Variants |
Length of simulation (ns) |
Number of conformers sampleda |
Number of microstates |
% population in specific microstates |
| 1 |
2 |
3 |
| Conformers sampled at 5 ps interval. |
| A1 |
10 |
2000 |
9 |
87.7 |
5.80 |
4.50 |
| A2 |
10 |
2000 |
5 |
96.2 |
2.20 |
1.50 |
| A3 |
10 |
2000 |
10 |
74.5 |
10.6 |
9.30 |
| A4 |
10 |
2000 |
7 |
73.3 |
18.2 |
4.30 |
| A5 |
10 |
2000 |
5 |
63.3 |
20.1 |
16.2 |
 |
| | Fig. 3 Ribbon diagram of central members of top microstate populated during molecular dynamics simulation of peptide variants. The aromatic residues (Tyr1, Phe5 and Trp16) for substrate binding and catalytic residues (Asp7, His10 and Ser12) for substrate hydrolysis are shown as purple and brown sticks, respectively. The percentage population in the top microstate is shown in parenthesis. | |
Table 3 Hydrogen bond distribution over complete molecular dynamics trajectory and statistics of radius of gyration (nm) over molecular fold (overall) and main chain atoms of peptide variants
| Variants |
Type |
Hydrogen bond statistics |
Radius of gyration (nm) |
| Avg./conf. |
Hydrogen bondsa |
| % SR |
% MR |
% LR |
| The hydrogen bonds are short (SR; i → i ± 2), medium (MR; i → i ± 3 + i → i ± 4) and long ranged (LR; i → i ± 5 + i → i ± ≥6). |
| A1 |
Overall |
7.4 |
3.6 |
24.1 |
72.3 |
0.72 ± 0.02 |
| Main chain |
6.2 |
2.0 |
18.2 |
79.8 |
0.66 ± 0.02 |
| A2 |
Overall |
6.8 |
3.0 |
27.6 |
69.4 |
0.70 ± 0.01 |
| Main chain |
5.1 |
2.9 |
26.1 |
71.0 |
0.65 ± 0.02 |
| A3 |
Overall |
6.6 |
4.8 |
18.4 |
76.8 |
0.74 ± 0.05 |
| Main chain |
5.7 |
3.7 |
17.9 |
78.4 |
0.70 ± 0.05 |
| A4 |
Overall |
7.5 |
5.9 |
20.0 |
74.2 |
0.69 ± 0.02 |
| Main chain |
4.7 |
8.3 |
18.9 |
72.9 |
0.61 ± 0.03 |
| A5 |
Overall |
7.1 |
5.6 |
20.8 |
73.6 |
0.76 ± 0.03 |
| Main chain |
5.9 |
2.8 |
18.9 |
78.3 |
0.70 ± 0.03 |
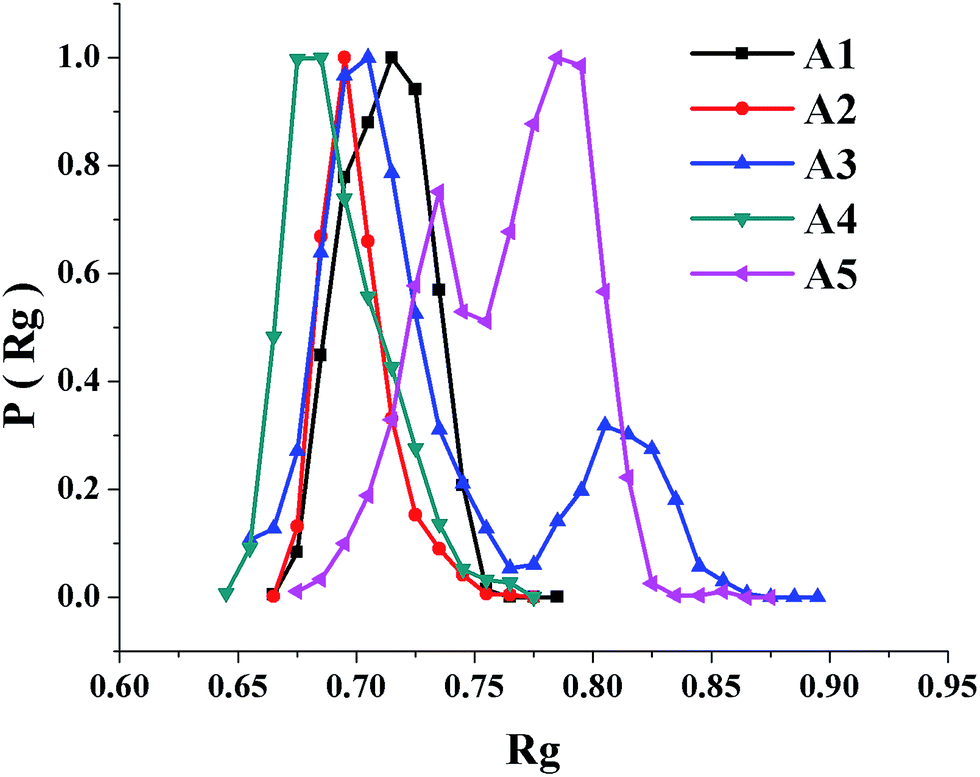
The MD ensembles were evaluated for microstates of the polypeptide structures. The statistics are summarized in Table 2 and the central members of the most populous microstates are shown in Fig. 3. The number of microstates populating the ensembles is ranged from 5 to 10. The most populous microstates are ranged from being 63% to 96% of the total ensemble. The polypeptides are remarkably ordered as specific microstates in concordance with our experiment data; convergence of favorable interactions involving both main chain and side chains may explain the result. Radius-of-gyration (Rg) distribution was evaluated over the conformers populating the MD ensembles. The observed Rg distributions are conformed to appreciable ordering of the designed sequences as β-hairpin folds. The variation of Rg distribution, except for A5, is similar for the peptides (Fig. 4).
 |
| | Fig. 4 Radius of gyration (Rg) distribution of the sixteen-residue peptide folds. Y-axis represents distribution of Rg over the conformers sampled during molecular dynamics simulation and X-axis represents Rg in nm. | |
Binding with p-nitrophenyl phosphate
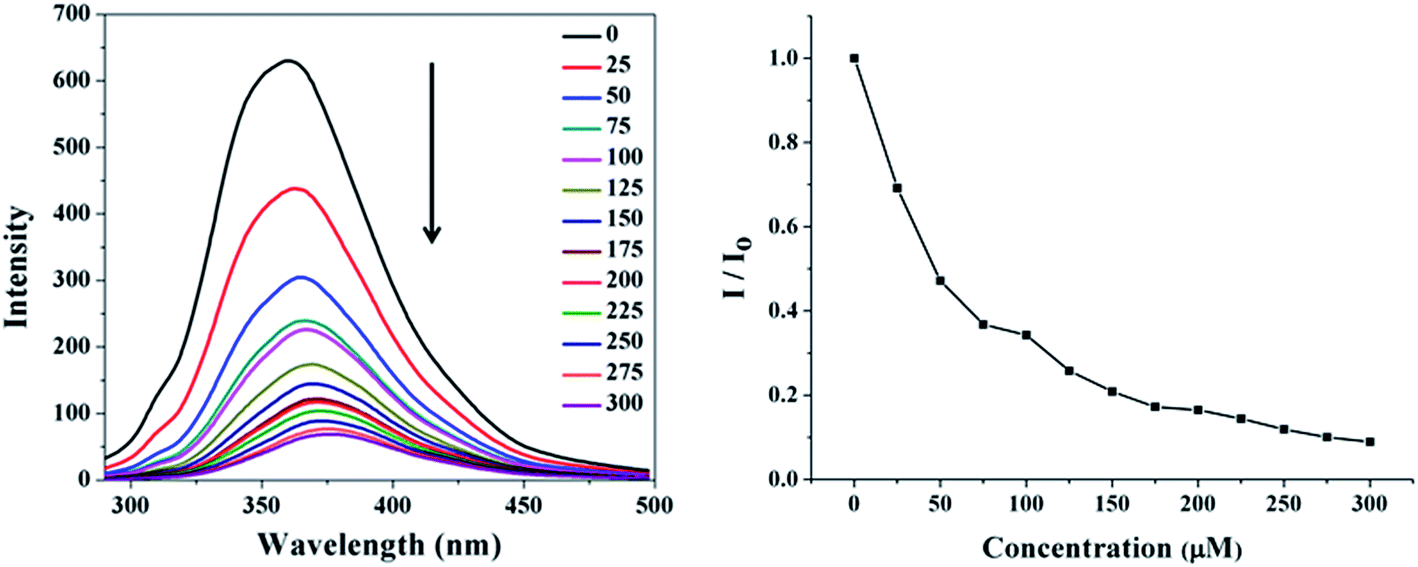
All peptides feature Trp as the potential fluorophore, which may vary in its environment according to sequence of the peptides. p-Nitrophenyl phosphate (pNPP) is a transition state analog for the hydrolysis reaction of pNPA; thus potentially it will be an inhibitor of the cognate esterase enzyme. The designed polypeptides were evaluated in affinity for pNPP with spectrofluorometry. All the sequences on titration with pNPP manifested quenching of Trp fluorescence (Fig. 5 and S4†); a ∼30 nm red shift of emission occurs on interaction of the peptides with pNPP. The binding energies of pNPP determined from the fluorescence quenching data are reported in Table 4. The binding energies are ranged from −7.21 to −8.64 kcal mol−1.
 |
| | Fig. 5 Quenching of tryptophan fluorescence of peptide A1 (15 μM) in sodium phosphate buffer (pH 7.0, 20 mM) at 298 K on progressive titration with increasing pNPP concentration (0–300 μM) (left panel), and plot of relative fluorescence intensity as a function of pNPP concentration (0–300 μM) (right panel). | |
Table 4 Binding energy of p-nitrophenyl phosphate (pNPP) with peptide variants determined with fluorescence and Michaelis–Menten kinetic parameters of peptide variants derived for the hydrolysis of p-nitrophenyl acetate (pNPA)
| Variants |
Binding energy (kcal mol−1) |
Michaelis–Menten kinetic parameters |
| 10−5 KM (M) |
10−5 kcat (s−1) |
kcat/KM (M−1 s−1) |
| A1 |
−7.21 ± 0.24 |
2.05 ± 0.58 |
6.58 ± 1.61 |
3.51 ± 1.56 |
| A2 |
−8.64 ± 0.57 |
1.55 ± 0.08 |
5.90 ± 1.10 |
3.85 ± 0.09 |
| A3 |
−7.89 ± 0.37 |
2.64 ± 1.62 |
6.93 ± 1.31 |
3.13 ± 1.31 |
| A4 |
−8.11 ± 0.28 |
1.85 ± 0.04 |
4.08 ± 0.01 |
2.21 ± 0.05 |
| A5 |
−8.14 ± 0.20 |
0.62 ± 0.04 |
4.95 ± 0.08 |
7.98 ± 0.67 |
Evaluation of catalysis
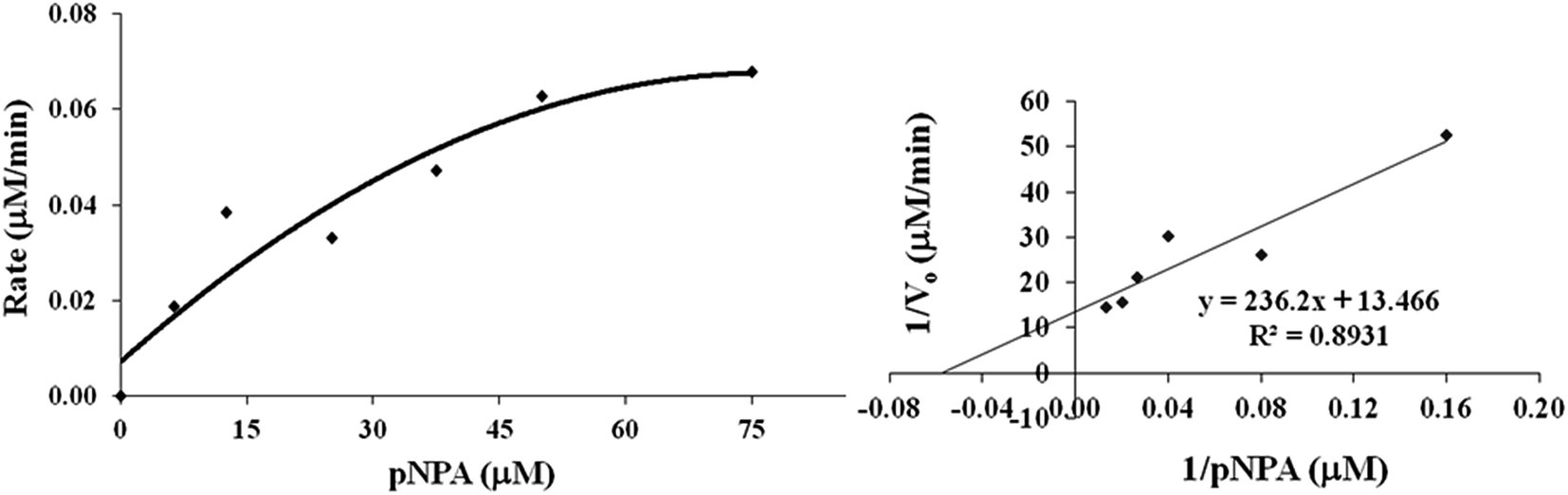
The designed sequences are evaluated for hydrolase activity against p-nitrophenyl acetate (pNPA) as the model ester substrate. The hydrolysis is easy to monitor as the reaction product is phenolate anion that is a strong chromophore. Michaelis–Menten kinetic parameters extracted for the hydrolysis reaction are presented in Table 4. Each synthetic peptide provides a clear evidence of saturation kinetics, being the hallmark of a real enzyme (Fig. 6 and S5†). All the polypeptides, except A5, are nearly comparable in catalytic efficiency and thus the programmed variations of structure are only marginal in the effect on catalytic power of the enzyme. A5 manifests slightly greater efficiency as a catalyst than A1. Possibly, the enhancement of flexibility of bent β-hairpin, by mutating the stereochemically rigid DPro8 with the less rigid DAsn8, improve catalytic efficiency of the enzyme. A2, having Glu as an acid group, manifests marginally higher catalytic efficiency than A1. A4, with aromatic and catalytic side chains mutually interchanged, manifests slightly lower catalytic efficiency than A1; specific orientation of the catalytic side chains, which are far off from the binding pocket of aromatic side chains (Tyr1, Phe5 and Trp16) could be important. Indeed, molecular dynamics simulation of A4 indicates that in the most populous microstate of the structure, the aromatic binding pocket and catalysis center of functional side chains are far apart from each other.
 |
| | Fig. 6 UV monitored rate of hydrolysis of pNPA with peptide A1 in sodium phosphate buffer (pH 7.0, 20 mM) at 298 K on progressive titration with increasing pNPA concentration (0–75 μM) in the form of Michaelis–Menten plot (left panel) and Lineweaver–Burk plot (right panel). | |
Discussion
The study was aimed to design a minimal hydrolase. Stepwise programming, first of a stereochemically defined fold and then of a chemically defined enzyme, was implemented. The fold was designed by exploiting our reported stereochemical approach;12 canonical poly-L β-hairpin was mutated stereochemically to suppress its intrinsic aggregation tendency and for its promotion as a monomolecular fold. The stereochemical mutation of poly-L β-hairpin, in a pair of cross-strand residues of L-structure to D-stereochemistry, promoted a ∼90° bend to produce a concave pocket in the naturally flat structure. The structure was elaborated chemically to create the desired specificity of ligand binding with aromatic side chains and enzyme activity with the appropriate functional side chains. Aromatic side chains were harnessed for stabilizing the fold besides providing the binding site for desired ligands on basis of π–π interactions, and functional side chains were harnessed for promotion of the binding site as a enzyme catalytic site for possible hydrolysis of an anchored substrate. The polypeptides were tested in the binding ability with pNPP as the reporter ligand and in the catalytic ability with pNPA as the reporter substrate. Chromogenicity of substrate offers convenience for possible future efforts to further evolve the catalyst. As a potential transition state analog for hydrolysis reaction of pNPA, pNPP was the surrogate ligand used for having the putative enzyme tested in its binding strength as a receptor protein.
The designed sequences were tested for specificity of both binding and catalysis with pNPP and pNPA as the reporter structures. The aromatic side chains were tested in the ability to fold the enzyme; exciton-coupled CD bands appearing in 220–230 nm range evidenced that Tyr1, Phe5 and Trp16 side chains indeed may be interacting as an aromatic cluster; the cluster was evidenced directly with MD. The clustering of the side chains occupying terminal positions of the polypeptide sequences conforms to ordering of the sequences as the desired hairpins. The hairpin and its nucleating β-turn were evidenced directly with MD and were noted to be locked with the required combination of long- and short-ranged hydrogen bonds in the main chain. Quenching of tryptophan fluorescence evidenced interaction of the designed fold with pNPP as the binding ligand. The appearance of phenolate anion observed with UV evidenced catalyzed hydrolysis of pNPA. The reactions were catalyzed with slower kinetics than with the typical natural enzymes (kcat/KM in the range 103 to 108 M−1 s−1)45 but were comparable to those of other enzymes of de novo design (kcat/KM in the range 10−2 to 102 M−1 s−1).6,46–55 kcat/KM varied narrowly, but the variations did evidence the favorable role of some of our specific design considerations. DPro in the sequence position 8 was replaced with DAsn; the consequent mobilization of β-hairpin as a fold accompanied its enhanced catalytic power as a hydrolase. Conformational mobility of enzymes is a well-documented requirement for their catalytic efficiency to accommodate the conformational requirements of catalytic turnover.56–60 The improved efficiency of the possible more flexible enzyme in A5 than in A1 is thus noteworthy. Compared to A1, A4 is lower in catalytic efficiency but is higher in binding affinity for pNPP. The weaker activity of A4 as an enzyme may be due to altered location of the functional side chains relative to aromatic binding pocket in the structure. In MD simulations, indeed the most populous microstate of A4 has its aromatic binding pocket far off from the cluster of functional side chains. The specific placement of aromatic side chains in the structure may explain why A4 has stronger affinity for pNPP than A1.
Although poor in catalytic efficiency, our hydrolase mimics are remarkable in their structural simplicity as polypeptide enzymes. Having been built from scratch by the stepwise approach, the mimics are the ideal models for possible further evolution and to have the underlying effects investigated with rigor. Stereochemistry has been further justified as the variable to diversify folds and to have them designed as minimal proteins that may be programmed for the desired function specificity chemically.
Conclusion
We described design of a stereochemically-bent sixteen-residue β-hairpin as a hydrolase mimic. In a stepwise approach, first the desired fold was programmed stereochemically, and then the desired sequence was programmed to the desired binding and catalysis specificity chemically. The binding measurements with a phophoester ligand and the rate measurements with a cognate carboxyester substrate have validated some of the design considerations we explored in the study. The accomplished hydrolases though modest in catalytic efficiency are remarkably simple polypeptide structures; the structures are promising for the possibility of future evolution by iterating structural changes and have the bases of possible improvements investigated with rigor.
Materials and methods
Materials
Fmoc-protected amino acids, reagents for solid-phase peptide synthesis, Rink-Amide AM resin, p-nitrophenyl acetate, and p-nitrophenyl phosphate was purchased from Sigma-Aldrich or Novabiochem-Merck.
Peptide design, synthesis and characterization
The peptide fold was modeled with the in-house software package CAPM (Computer Aided Peptide Modeling),12 capable of handling D-amino acids effectively. In-house program PDBmake was used to generate coordinates of the CAPM modeled structure. The peptide sequences were designed with help of in-house sequence optimization program IDeAS, capable of handling D-amino acid effectively. The peptides were synthesized manually on Rink-Amide AM resin using standard Fmoc chemistry and HOBt/DIC as the coupling reagent.61 The coupling reaction was monitored with standard Kaiser and chloranil tests, each coupling reaction typically required about 6 h. Deprotections were carried out with 30% (v/v) piperidine–DMF. N-terminus was acetylated (–NHCOCH3) with Ac2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DIPEA:DMF in 1:2:20 ratio. Cleavage of the final polypeptide and deprotection of side chains were achieved together with reagent K (82.5% TFA/5% dry-phenol/5% thioanisole/2.5% ethandithiol/5% water). The product precipitated with anhydrous diethyl ether was lyophilized from H2O:tBuOH (1 : 4) solution as a white powder. Mass spectra of the synthesized peptides were recorded on QTOF-ESI mass spectrometer. Positive ions were detected in linear/reflectron mode. 1H NMR spectra were recorded on 700 MHz Bruker instrument at 298 K. Peptide concentrations of 2.5 mM were used. 1H NMR spectra were recorded for all peptides at 10-fold dilutions to check the formation of aggregation. Solutions were prepared in 90% H2O/10% D2O with 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as the internal reference. Solvent was suppressed with pre-saturation or WATER suppression by GrAdient Tailored Excitation (WATERGATE) sequence, as provided in the Bruker software.
:DIPEA:DMF in 1:2:20 ratio. Cleavage of the final polypeptide and deprotection of side chains were achieved together with reagent K (82.5% TFA/5% dry-phenol/5% thioanisole/2.5% ethandithiol/5% water). The product precipitated with anhydrous diethyl ether was lyophilized from H2O:tBuOH (1 : 4) solution as a white powder. Mass spectra of the synthesized peptides were recorded on QTOF-ESI mass spectrometer. Positive ions were detected in linear/reflectron mode. 1H NMR spectra were recorded on 700 MHz Bruker instrument at 298 K. Peptide concentrations of 2.5 mM were used. 1H NMR spectra were recorded for all peptides at 10-fold dilutions to check the formation of aggregation. Solutions were prepared in 90% H2O/10% D2O with 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as the internal reference. Solvent was suppressed with pre-saturation or WATER suppression by GrAdient Tailored Excitation (WATERGATE) sequence, as provided in the Bruker software.
Circular dichroism
Circular Dichroism (CD) was recorded on a JASCO J-180 CD spectropolarimeter calibrated with d10-camphorsulphonic acid. CD spectra were recorded at 298 K in a 0.2 cm path length quartz cell with a 2 nm bandwidth in the far-UV (190–250 nm) range. Scanning at 100 nm min−1 with 1.0 s time constant in 1 nm steps, five scans were averaged after baseline correction for water. Working solutions of 20–100 μM concentration of peptide were prepared by optical measurements. The observations in millidegrees were converted to mean residue molar ellipticity [θMRW].
Spectrofluorometry
The quenching of Trp fluorescence was measured on a Perkin Elmer LS-55 spectrofluorimeter equipped with a standard PMT. Data were collected at 298 K in 1 ml cell, with λexcitation as 295 nm, λemission in the 300–450 nm range with 2.5 nm excitation and emission slits. Scanning at 100 nm min−1 with 1 nm steps was used. The working concentrations 15 μM concentration of peptide and N-acetyl-L-trytophanamide (NATA) were calibrated by OD measurements using molar extinction coefficient of Trp (∼5600 at 280 nm) and Tyr (∼1280 at 280 nm). Binding experiments were carried out at 15 μM concentration of peptide and 0–300 μM concentration of pNPP. Stern–Volmer constants (KSV) for the external quencher i.e. pNPP, were obtained using the following biomolecular quenching equation:
where Io = fluorescence intensity in the absence of external quencher, I = fluorescence intensity in the presence of quencher, Q = concentration of the quencher, and KSV = Stern–Volmer constant calculated from the slope of line. The emission maximum intensities of tryptophan were fit as a function of pNPP concentration to the described 1:1 binding isotherm, and Kd, hence binding energy, was estimated.
Enzyme activity
The kinetics of hydrolysis was monitored spectrophotometrically on a Perkin Elmer spectrophotometer using p-nitrophenyl acetate (pNPA) as substrate, by observing the production of the p-nitrophenolate ion at 410 nm.62 A stock solution of pNPA was prepared in water with a few drops of acetonitrile added to solubilize pNPA. Peptide concentration in the assays was in the range of 20 μM. Hydrolase activity was evaluated in 20 mM phosphate buffer of pH 7.0 at 298 K, by varying substrate pNPA concentration (0–75 μM). The catalyzed rate of pNPA hydrolysis was measured by an initial slope method, following the increase in 410 nm absorption by the p-nitrophenolate ion.
Molecular dynamics
Molecular dynamics was performed with gromos-96 43A1 force field in GROMACS (GROningen MAchine for Chemical Simulations) package63,64 in a periodic box of explicit solvent using water. The simulation was performed under NVT condition, viz., fixed number of particles, constant volume, and constant temperature. Non-bonded list cutoff was 1.4 nm with shift at 0.8 nm. Integration step was 2 fs. Initial velocities were drawn from Maxwellian distribution. Temperature was coupled to an external bath with relaxation time constant of 0.1 ps. Bond lengths were constrained with SHAKE65 to geometric accuracy 10−4. Peptides constrained to center of the periodic cubic box were soaked in SPC (Simple Point Charge) water,66 which was added to 1 atm density at 298 K. For an electrically charged system, counter ions were added by replacing the solvent molecules to achieve electrical neutrality. Electrostatics was in this case treated with the Particle Mesh Ewald (PME)67,68 method implementing a Coulomb cutoff of 1.4 nm, Fourier spacing of 0.12 nm, and an interpolation order of 4. First solute was energy minimized, and then solvent was energy minimized while restraining solute, and finally both were energy minimized after removing restraint. Simulation was initialized and 3 ns trajectory was exempted from analysis as pre-equilibration period. The trajectory was sampled thereafter at 5 ps intervals. To achieve the microstates of peptide structure conformational clustering to 0.15 nm rmsd cutoff was performed with a reported procedure.69
Acknowledgements
We acknowledge Department of Science and Technology (09DST028), Government of India for the financial support and IIT Bombay for the computing facility “Corona”.
References
- B. I. Dahiyat and S. L. Mayo, Science, 1997, 278, 82–87 CrossRef CAS PubMed.
- J. R. Desjarlais and T. M. Handel, Protein Sci., 1995, 4, 2006–2018 CrossRef CAS.
- J. W. Ponder and F. M. Richards, J. Mol. Biol., 1987, 193, 775–791 CrossRef CAS.
- J. P. Schneider, A. Lombardi and W. F. Degrado, Folding Des., 1998, 3, R29–R40 CrossRef CAS PubMed.
- H. W. Hellinga and F. M. Richards, J. Mol. Biol., 1991, 222, 763–785 CrossRef CAS PubMed.
- D. N. Bolon and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14274–14279 CrossRef CAS PubMed.
- H. W. Hellinga and F. M. Richards, J. Mol. Biol., 1991, 222, 787–803 CrossRef CAS.
- J. Reina, E. Lacroix, S. D. Hobson, G. Fernandez-Ballester, V. Rybin, M. S. Schwab, L. Serrano and C. Gonzalez, Nat. Struct. Biol., 2002, 9, 621–627 CAS.
- L. L. Looger, M. A. Dwyer, J. J. Smith and H. W. Hellinga, Nature, 2003, 423, 185–190 CrossRef CAS PubMed.
- S. M. Malakauskas and S. L. Mayo, Nat. Struct. Biol., 1998, 5, 470–475 CrossRef CAS PubMed.
- P. Koehl and M. Levitt, J. Mol. Biol., 1999, 293, 1161–1181 CrossRef CAS.
- S. Durani, Acc. Chem. Res., 2008, 41, 1301–1308 CrossRef CAS PubMed.
- S. Rana, B. Kundu and S. Durani, Bioorg. Med. Chem., 2007, 15, 3874–3882 CrossRef CAS PubMed.
- S. Rana, B. Kundu and S. Durani, Biopolymers, 2007, 87, 231–243 CrossRef CAS PubMed.
- K. Patel, B. Goyal, A. Kumar, N. Kishore and S. Durani, J. Phys. Chem. B, 2010, 114, 16887–16893 CrossRef CAS PubMed.
- K. Patel, K. R. Srivastava and S. Durani, Bioorg. Med. Chem., 2010, 18, 8270–8276 CrossRef CAS PubMed.
- R. Sterner and F. X. Schmid, Science, 2004, 304, 1916–1917 CrossRef CAS PubMed.
- M. D. Ballinger, J. Tom and J. A. Wells, Biochemistry, 1995, 34, 13312–13319 CrossRef CAS PubMed.
- V. Nanda, Nat. Chem. Biol., 2008, 4, 273–275 CrossRef CAS.
- V. Nanda and R. L. Koder, Nat. Chem., 2010, 2, 15–24 CrossRef CAS PubMed.
- C. M. Tann, D. Qi and M. D. Distefano, Curr. Opin. Chem. Biol., 2001, 5, 696–704 CrossRef CAS.
- D. N. Woolfson, G. J. Bartlett, A. J. Burton, J. W. Heal, A. Niitsu, A. R. Thomson and C. W. Wood, Curr. Opin. Struct. Biol., 2015, 33, 16–26 CrossRef CAS.
- K. Świderek, I. Tuñón, V. Moliner and J. Bertran, Arch. Biochem. Biophys., 2015, 582, 68–79 CrossRef PubMed.
- S. Rämisch, U. Weininger, J. Martinsson, M. Akke and I. André, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17875–17880 CrossRef.
- D. Suplatov, N. Panin, E. Kirilin, T. Shcherbakova, P. Kudryavtsev and V. Švedas, PLoS One, 2014, 9, e100643 Search PubMed.
- J. Damborsky and J. Brezovsky, Curr. Opin. Chem. Biol., 2014, 19, 8–16 CrossRef CAS PubMed.
- H. J. Wijma and D. B. Janssen, FEBS J., 2013, 280, 2948–2960 CrossRef CAS PubMed.
- N. Kobayashi, K. Yanase, T. Sato, S. Unzai, M. H. Hecht and R. Arai, J. Am. Chem. Soc., 2015, 137, 11285–11293 CrossRef CAS PubMed.
- H. J. Wijma, R. J. Floor, P. A. Jekel, D. Baker, S. J. Marrink and D. B. Janssen, Protein Eng., Des. Sel., 2014, 27, 49–58 CrossRef CAS PubMed.
- S. Rajagopalan, C. Wang, K. Yu, A. P. Kuzin, F. Richter, S. Lew, A. E. Miklos, M. L. Matthews, J. Seetharaman, M. Su, J. F. Hunt, B. F. Cravatt and D. Baker, Nat. Chem. Biol., 2014, 10, 386–391 CrossRef CAS PubMed.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS PubMed.
- J. A. Stapleton, T. A. Whitehead and V. Nanda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9632–9637 CrossRef CAS PubMed.
- E. Straucha, S. J. Fleishmana and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 675–680 CrossRef PubMed.
- T. Heinisch, M. Pellizzoni, M. Dürrenberger, C. E. Tinberg, V. Köhler, J. Klehr, D. Häussinger, D. Baker and T. R. Ward, J. Am. Chem. Soc., 2015, 137, 10414–10419 CrossRef PubMed.
- H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 1998, 120, 4236–4237 CrossRef CAS.
- M. Harel, D. M. Quinn, H. K. Nair, I. Silman and J. L. Sussman, J. Am. Chem. Soc., 1996, 118, 2340–2346 CrossRef CAS.
- A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5578–5583 CrossRef CAS PubMed.
- N. H. Andersen, K. A. Olsen, R. M. Fesinmeyer, X. Tan, F. M. Hudson, L. A. Eidenschink and S. R. Farazi, J. Am. Chem. Soc., 2006, 128, 6101–6110 CrossRef CAS PubMed.
- B. N. Markiewicz, R. Oyola, D. Du and F. Gai, Biochemistry, 2014, 53, 1146–1154 CrossRef CAS PubMed.
- A. Abelein, J. P. Abrahams, J. Danielsson, A. Gräslund, J. Jarvet, J. Luo, A. Tiiman and S. K. Wärmländer, J. Biol. Inorg. Chem., 2014, 19, 623–634 CrossRef CAS PubMed.
- C. J. Bowerman, W. Liyanage, A. J. Federation and B. L. Nilsson, Biomacromolecules, 2011, 12, 2735–2745 CrossRef CAS PubMed.
- K. Kar, C. L Hoop, K. W. Drombosky, M. A. Baker, R. Kodali, I. Arduini, P. C. van der Wel, W. S. Horne and R. Wetzel, J. Mol. Biol., 2013, 425, 1183–1197 CrossRef CAS PubMed.
- L. Luca and J. E. Shea, Biophys. J., 2012, 103, 576–586 CrossRef PubMed.
- A. C. Gill, PLoS One, 2014, 9, e87354 Search PubMed.
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi, D. S. Tawfik and R. Milo, Biochemistry, 2011, 50, 4402–4410 CrossRef CAS.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS PubMed.
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190–195 CrossRef PubMed.
- H. K. Privett, G. Kiss, T. M. Lee, R. Blomberg, R. A. Chica, L. M. Thomas, D. Hilvert, K. N. Houk and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3790–3795 CrossRef CAS PubMed.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas III, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- E. A. Althoff, L. Wang, L. Jiang, L. Giger, J. K. Lassila, Z. Wang, M. Smith, S. Hari, P. Kast, D. Herschlag, D. Hilvert and D. Baker, Protein Sci., 2012, 21, 717–726 CrossRef CAS PubMed.
- S. Bjelic, L. G. Nivón, N. Çelebi-Ölçüm, G. Kiss, C. F. Rosewall, H. M. Lovick, E. L. Ingalls, J. L. Gallaher, J. Seetharaman, S. Lew, G. T. Montelione, J. F. Hunt, F. E. Michael, K. N. Houk and D. Baker, ACS Chem. Biol., 2013, 8, 749–757 CrossRef CAS PubMed.
- J. K. Lassila, D. Baker and D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4937–4942 CrossRef CAS PubMed.
- I. V. Korendovych, D. W. Kulp, Y. Wu, H. Cheng, H. Roder and W. F. Degrado, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6823–6827 CrossRef CAS PubMed.
- A. L. Pinto, H. W. Hellinga and J. P. Caradonna, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 5562–5567 CrossRef CAS.
- M. Suarez, P. Tortosa, M. M. Garcia-Mira, D. Rodríguez-Larrea, R. Godoy-Ruiz, B. Ibarra-Molero, J. M. Sanchez-Ruiz and A. Jaramillo, Biophys. Chem., 2010, 147, 13–19 CrossRef CAS PubMed.
- A. Bar-Even, R. Milo, E. Noor and D. S. Tawfik, Biochemistry, 2015, 54, 4969–4977 CrossRef CAS PubMed.
- M. Kokkinidis, N. M. Glykos and V. E. Fadouloglou, Adv. Protein Chem. Struct. Biol., 2012, 87, 181–218 CAS.
- N. Tokuriki and D. S. Tawfik, Science, 2009, 324, 203–207 CrossRef CAS PubMed.
- E. Z. Eisenmesser, O. Millet, W. Labeikovsky, D. M. Korzhnev, M. Wolf-Watz, D. A. Bosco, J. J. Skalicky, L. E. Kay and D. Kern, Nature, 2005, 438, 117–121 CrossRef CAS PubMed.
- B. X. Yan and Y. Q. Sun, J. Biol. Chem., 1997, 272, 3190–3194 CrossRef CAS PubMed.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, USA, 2000 Search PubMed.
- O. A. Bessey and R. H. Love, J. Biol. Chem., 1952, 196, 175–178 CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CAS.
- W. F. van Gunsteren, S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott and I. G. Tironi, Biomolecular Simulation: The GROMOS96 Manual and User Guide, Vdf Hochschulverlag AG an der ETH Zürich, Zürich, Switzerland, 1996, pp. 1–1042 Search PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction models for water in relation to protein hydration, Intermolecular Forces, ed. B. Pullman, Reidel Publishing Company: Dordrecht, The Netherlands, 1981, pp. 331–342 Search PubMed.
- T. Darden, D. York and L. J. Pedersen, Chem. Phys., 1993, 98, 10089–10092 CAS.
- U. Essmann, M. L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The data of mass, NMR, CD, fluorescence and kinetics studies can be found. Fig. S1–S5. See DOI: 10.1039/c5ra19015k |
| ‡ Present address Department of Chemistry, School of Basic and Applied Sciences, Sri Guru Granth Sahib World University, Fatehgarh Sahib–140406, Punjab, India. |
| § Department of Chemistry, N. B. Mehta Science College, Bordi, Dahanu, Dist. Thane, Maharashtra, India. |
| ¶ Department of Physics and Astronomy, Michigan State University, East Lansing, USA. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Kirti Patel§
,
Kinshuk Raj Srivastava¶
and
Susheel Durani*
*,
Kirti Patel§
,
Kinshuk Raj Srivastava¶
and
Susheel Durani*