
Integrated analysis of serum and intact muscle metabonomics identify metabolic profiles of cancer cachexia in a dynamic mouse model†
Abstract
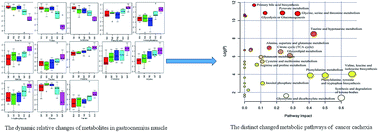
Cancer cachexia is a multifactorial metabolic syndrome characterized by a severe loss of body weight and lean body mass. Metabolic dysfunction is the primary hallmark of muscle atrophy. Herein, we studied dynamic metabolic profiles in serum and intact muscle. High-resolution magic angle spinning was employed for intact gastrocnemius muscle analysis and a dynamic metabolic model was established using C26 colon carcinoma-bearing mice from procachexia to the refractory cachexia period. When an integrated analysis of the 13 metabolites from the intact muscle gastrocnemius and 43 metabolites from the serum was performed, five distinguishable metabolic features were identified, including low blood glucose, elevated ketone bodies, decreased branched-chain amino acids, increased neutral amino acids, and high 3-methylhistidine and creatine. The metabolic hubs reveal potential biomarkers for the early detection of cachexia and indicate the underlying metabolic pathway reprogramming of muscle atrophy.
 Please wait while we load your content...
Please wait while we load your content...

