
Kaolinite properties and advances for solid acid and basic catalyst synthesis
Peter Adeniyi Alaba
a,
Yahaya Muhammad Sani
ab and
Wan Mohd Ashri Wan Daud
*a
aDepartment of Chemical Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: ashri@um.edu.my; adeniyipee@live.com; ymsani@abu.edu.ng; Fax: +60 3 79675319; Tel: +60 1 126376195 Tel: +60 3 79675297
bDepartment of Chemical Engineering, Ahmadu Bello University, 870001 Nigeria
First published on 18th November 2015
Abstract
Historically, clay mineral catalysts have found industrial applications since the early 1930s. However, inherent limitations such as impurities, porosity, low surface area and acidity hindered their wide and sustained acceptability, this is despite their economic advantages. Interestingly, the use of kaolinite as precursor in active catalyst synthesis has been a breakthrough for several industrial processes such as petrol chemistry; especially in catalytic refining and bulk chemistry. The same is also true for processes that require solid acid catalysts, catalyst support, co-catalyst or promoter application for positive environmental impact and economic viability. Therefore, this article reviews the physicochemical properties of kaolinite and their amenability to modification towards enhancing their catalytic properties. The article also discussed modification methods such as mechanochemical activation (dealumination), thermal activation, intercalation and chemical activation. With more advances in technology and long-term commitment to investments, kaolinite will become the ideal catalyst and precursor for synthesizing novel catalysts for a sustainable “greener” future.
 Peter Adeniyi Alaba | Peter Adeniyi Alaba was born in Kwara State, Nigeria in 1980 and carried out undergraduate at the Federal University of Technology Minna. He is currently concluded his postgraduate studies (Mphil.) at the University of Malaya under the supervision of Prof. Wan Mohd Ashri Wan Daud. He was appointed to a Graduate research assistantship at the University of Malaya in 2013 till date. His research interests lie in rational design of tailored solid acid catalysts for efficient biofuel production, environmental engineering, CFD, and computational chemistry. |
 Yahaya Muhammad Sani | Yahaya Muhammad Sani was born and raised in Zaria, a city rich in cultural heritage & the capital of the Hausa kingdom of Zazzau in Nigeria. He received his B.Eng. Chem. Eng. from Ahmadu Bello University, the 2nd largest on the African continent (2002), MSc degrees in Biotechnology (Newcastle University, UK; 2007), & Chem. Eng. (A.B.U. Zaria; 2008) prior to obtaining a PhD from University of Malaya, Malaysia. He joined A.B.U. Zaria in 2005 with research interest on catalysis, and renewable & sustainable energy. He is a registered Engineer certified by Council for the Regulation of Engineering, & a corporate member of the Nigerian Society of Engineers. |
 Wan Mohd Ashri Wan Daud | Professor Wan Mohd Ashri Wan Daud received his bachelor degree in Chemical Engineering at Leeds University, Leeds, UK in 1991 and his master's degree in Chemical Engineering at the University of Sheffield, Sheffield, UK in 1993. He received his PhD degree in Chemical Engineering at the University of Sheffield in 1996. After nine years as an academic and scientist at the Faculty of Engineering, in 2005, he became Professor of Chemical Engineering. Since 2005 until now, he has worked as a Professor of Chemical Engineering at the University of Malaya, Malaysia. His research fields include energy, biomass conversion to bio-fuel, catalysts synthesis, polymerization and separation processes, and hydrogen storage. He has more than 131 publications in Web Science journals. |
1. Introduction
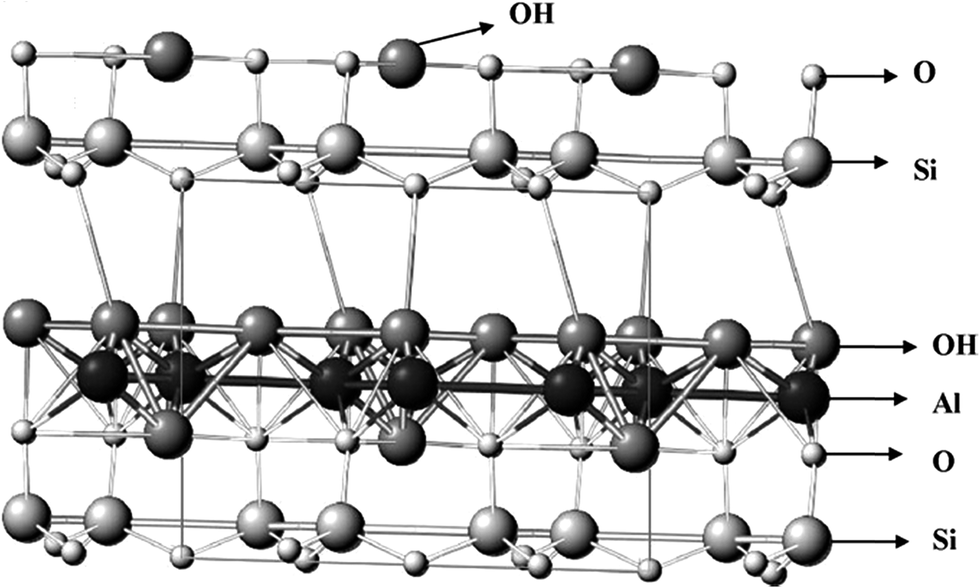
Kaolinite is a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 layer phyllosilicate clay mineral that makes up about 10–95% of the mineral kaolin. Its structure possesses a sheet of gibbsite-like Al(OH)4 (octahedrally coordinated) and a sheet of SiO4 tetrahedral combined with longitudinal sideline chains forming a dioctahedral structure called 1:1 layer phyllosilicate.1–3 Fig. 1 shows the structural model of kaolinite.4 Theoretically, its chemical formula is Si2Al2O5 (OH)4 (Al2O5 Si2·2H2O or Al2O3·2SiO4·2H2O).5–7 Other components of kaolin includes mica and quartz as well as metal oxides such as K2O, CaO, TiO2, Fe2O3, Na2O, MgO, MnO, and P2O5 as impurities. It is a versatile hydrated aluminosilicate with a wide variety of industrial applications such as in catalysis, photocatalysis,8 ion exchange,9 decolorization,10–12 adsorption,12,13 catalyst supports and modified catalysts.14 However, despite their economic advantages, kaolinite materials need to be modified because of their inherent limitations such as impurities, porosity, low surface area and acidity which hindered their wide and sustained acceptability. Modified kaolinite catalysts and supports are regaining attention in catalysis because they are naturally abundant.15,16 Other favorable factors include their improved surface area, mechanical and chemical stability, and high Lewis and Brønsted acid sites density. The materials are also environmentally benign9,17 with cation exchange capacity (CEC). Reactivity and surface properties have enhanced the application of kaolinite in the chemical industry based on surface modifications. There are various methods, as proposed in the open literature, for enhancing the properties of kaolinite. These include chemical activation (intercalation, alkali, organic and inorganic acid),17 intercalation,18 mechanochemical,19,20 and thermal treatments.17,18
:1 layer phyllosilicate clay mineral that makes up about 10–95% of the mineral kaolin. Its structure possesses a sheet of gibbsite-like Al(OH)4 (octahedrally coordinated) and a sheet of SiO4 tetrahedral combined with longitudinal sideline chains forming a dioctahedral structure called 1:1 layer phyllosilicate.1–3 Fig. 1 shows the structural model of kaolinite.4 Theoretically, its chemical formula is Si2Al2O5 (OH)4 (Al2O5 Si2·2H2O or Al2O3·2SiO4·2H2O).5–7 Other components of kaolin includes mica and quartz as well as metal oxides such as K2O, CaO, TiO2, Fe2O3, Na2O, MgO, MnO, and P2O5 as impurities. It is a versatile hydrated aluminosilicate with a wide variety of industrial applications such as in catalysis, photocatalysis,8 ion exchange,9 decolorization,10–12 adsorption,12,13 catalyst supports and modified catalysts.14 However, despite their economic advantages, kaolinite materials need to be modified because of their inherent limitations such as impurities, porosity, low surface area and acidity which hindered their wide and sustained acceptability. Modified kaolinite catalysts and supports are regaining attention in catalysis because they are naturally abundant.15,16 Other favorable factors include their improved surface area, mechanical and chemical stability, and high Lewis and Brønsted acid sites density. The materials are also environmentally benign9,17 with cation exchange capacity (CEC). Reactivity and surface properties have enhanced the application of kaolinite in the chemical industry based on surface modifications. There are various methods, as proposed in the open literature, for enhancing the properties of kaolinite. These include chemical activation (intercalation, alkali, organic and inorganic acid),17 intercalation,18 mechanochemical,19,20 and thermal treatments.17,18
 | ||
| Fig. 1 Shows the structural model of kaolinite.4 | ||
However, modified kaolinite have received much attention because of their crystalline structure21 and large pore size, which are advantageous in the conversion of bulky molecules.22 Applying mechanochemical techniques has great effects on surface modifications, which is pronounced in Szeg and Zettlitz kaolinite. Mechanochemical activation of these materials improves their active centers, surface area, and pore volume.20,23 Kaolinite materials are also amenable to alkali, organic and inorganic acid activation.
Acid and alkali treatment dissolve the external layer and eliminate impurities. Further, dealumination or desilication of kaolinite' structure changes the structural and chemical composition16 of the material. Similarly, the number of acid sites, surface area and catalytic properties of amorphized kaolin (calcined or grounded), smectic clays (montmorillonite and saponite) and fibrous clays (palygorskite and saponite)19,20,22,23 increases. Their wide range of pore sizes, strong acid sites and partially amorphous nature17,24 facilitate the strong acidic properties of modified kaolinite catalysts. These make them viable for dehydration reactions, biomass conversion,17 and catalytic cracking.10,17,25 Modified kaolinite may also act as meso- and macroporous supports for transition metal oxide adsorbents or catalysts in the reacting mixture because of their partially amorphous nature. Furthermore, modification improves the effectiveness,26,28 reactivity,11,28 and efficiency11,30 as well as stability11 of the catalysts via increased aggregation of metal oxide.11,30 Instances of such application include pollution control removal of impurities,30 especially in waste water treatment11 and use as a photo-Fenton catalyst.31 Others include free fatty acid (FFA) esterification17 and steam gasification.27 Interestingly, kaolinite are impervious to microbial attack, thermally stable, hydrophilic in nature and easily regenerated. Some authors have reported the suitability of kaolinite as inorganic supports for enzymatic (organic) catalysts.31–34 Furthermore, several researchers have highlighted these materials as economical precursors in synthesizing microspherical zeolitic molecular sieves such as Y and X zeolites. Evidently, zeolites are popular in fluid catalytic cracking processes.36–38
Despite extensive researches on catalytic applications of these materials, the advent of synthetic silica/alumina and zeolite Y has seemingly overshadowed their popularity. Moreover, rapidly expanding FCC units led to increased catalyst consumption. This created a dearth of high quality starting materials for in situ synthesis. Therefore, this review aims at exploring the physicochemical properties of kaolinite that make them suitable for catalysis. To achieve this aim seamlessly, the article reviewed the physicochemistry and how their amenability to modification enhances catalysis. The review comprises six sections: an introductory section, followed by a general discussion on physicochemical properties and modification in Section 2. Section 3 reviews the different types of modifications while Section 4 presents instances that discuss the applicability of kaolinite as a precursor in synthesizing microspherical zeolites. To stimulate further researches, Section 5 gives in-depth analyzes of kaolinite as catalyst supports while Section 6 highlights applications of these catalytic materials. The discussion concludes by proffering promising solutions that will ensure the prominence of kaolinite in catalysis. These include recent advances in improving the development of new materials.
2. Physicochemical properties
The physicochemical characteristics of kaolinite determine their industrial applications in catalysis. Manipulating these properties increases the industrial applicability of these materials by enhancing their activity and product selectivity.39 Therefore, this section highlights how analyzes such as XRD, TGA, DSC and FTIR reveal modifications in kaolinite. The section also discusses instances in which thermal activation and surface charge heterogeneity affect these physicochemical properties. Table 1 shows the chemical composition of different kaolin samples with Si/Al ratios ranging from 1.1 to 1.78. The table also highlights impurities such as K2O, CaO, TiO2, Fe2O3, Na2O, MgO, MnO, and P2O5.| Kaolinite sample | Si/Al | K2O (wt%) | CaO (wt%) | TiO2 (wt%) | Fe2O3 (wt%) | Na2O (wt%) | MgO wt(%) | MnO (wt%) | P2O5 (wt%) | H2O/LOIa (wt%) | d001 | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Notation | Origin | ||||||||||||
| a LOI: loss on ignition. | |||||||||||||
| AK | Unknown | 1.20 | 0.9 | — | 0.4 | 0.4 | — | — | — | — | — | 7.202 | 40 |
| FK | Brittany (F) | 1.30 | 2.5 | — | 0.5 | — | — | — | — | — | 7.110 | 40 | |
| EK | Unknown | 1.20 | 1 | — | 0.4 | 0.4 | — | — | — | — | — | 7.237 | 40 |
| DK | Charentes basin (F) | 1.10 | 0.5 | 0.4 | 0.7 | 0.8 | — | — | — | — | — | 7.231 | 40 |
| MK | Mahoming, China | 1.24 | 0.12 | 0.15 | 0.25 | 0.41 | 0.18 | 0.22 | — | — | 14.00 | 7.108 | 8 |
| BK | Beihai, China | 1.26 | 0.9 | 0.01 | 0.05 | 0.32 | 0.2 | 0.12 | — | — | 13.47 | 7.162 | 8 |
| HK | Hanpu, China | 1.34 | 1.26 | 0.01 | 0.06 | 0.39 | 0.03 | 0.15 | — | — | 12.51 | 7.108 | 8 |
| LK | Longyan, China | 1.24 | 0.19 | 0.1 | 0.12 | 0.74 | 0.03 | 0.04 | 0.01 | — | 14.68 | 7.108 | 8 |
| KK | Zhangjiakou, China | 1.30 | 0.01 | 0.47 | 1.38 | 0.4 | 0.08 | 0.01 | 0 | 0.1 | 13.93 | 7.170 | 41 |
| CK | Datong, China | 1.78 | 0.6 | 0.39 | 0.05 | 1.52 | 0.72 | 1.33 | 0.07 | <0.1 | 11.61 | 7.170 | 41 |
| KF | Damrec, France | 1.49 | 0.34 | 0.03 | 0.12 | 0.68 | 0.08 | 0.08 | 12.70 | 7.262 | 41 | ||
2.1. FTIR studies
Fourier transformed infrared (FTIR) techniques detect bonding, acidity, structural and chemical properties of kaolinite according to their vibrational mode.2,37–44 Generally, kaolinite exhibit structural hydroxyl group OH stretching vibration between 3703 and 3566 cm−1 and physically absorbed water is present around 1629 cm−1. The apical Si–O in-plane stretching vibration is between 1107 and 1118 cm−1, and the skeleton Si–O–Si in-plane stretching vibration is between 1014 and 1032 cm−1. The Al–OH bending vibration is between 912 and 938 cm−1, and the Al–OH translational vibration is between 695 and 796 cm−1. They also exhibit a Si–O–Al bending vibration between 536 and 541 cm−1 and Si–O bending vibrations at 426 and 471 cm−1. Table 2 shows this information for different kaolinite materials.8,23,25| Suggested assignments | Wave numbers (cm−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| MK | BK | HK | LK | AK | FK | EK | DK | |
| Si–OH stretching vibration | 3740 | — | — | 3739 | — | — | — | — |
| Inner surface –OH in-phase stretching (strong) vibration | 3695 | 3703 | 3701 | 3696 | 3695 | 3695 | 3696 | 3695 |
| Inner surface –OH out-phase stretching (medium) vibration | 3660 | 3650 | 3650 | 3661 | 3669 | 3669 | 3669 | 3669 |
| — | — | — | — | 3652 | 3652 | 3652 | 3653 | |
| Inner surface –OH stretching (strong) vibration | 3622 | 3619 | 3620 | 3620 | 3619 | 3619 | 3619 | 3619 |
| Inner layer water –OH vibration | — | — | — | 3566 | — | — | — | — |
| Physical absorbed water –OH vibration | 1629 | 1624 | 1624 | 1625 | — | — | — | — |
| Apical Si–O in-plane stretching vibration | 1107 | 1113 | 1118 | — | 1114 | 1114 | 1114 | 1112 |
| Skeleton Si–O–Si in-plane stretching vibration | 1014 | 1031 | 1032 | 1033 | 1031 | 1031 | 1031 | 1031 |
| Al–OH bending vibration | 918 | 912 | 912 | 913 | 938 | 938 | 938 | 936 |
| AL–OH (“gibbsite-like” layer) translational vibration | 792 | 795 | 796 | 794 | 792 | 792 | 792 | 794 |
| 755 | 755 | 754 | 754 | 755 | 755 | 756 | 755 | |
| 695 | 697 | 696 | 689 | 699 | 698 | 699 | 698 | |
| Si–O–Al bending vibration | 541 | 538 | 536 | 538 | 537 | 539 | 537 | 537 |
| Si–O bending vibration | 471 | 468 | 469 | 470 | 469 | 469 | 469 | 469 |
| 426 | 429 | 430 | 432 | 429 | 429 | 429 | 429 | |
2.2. XRD studies
XRD diffractograms reveal structural defects in kaolinite because of variability in the peak positions and modulation of their intensities in kaolinite XRD patterns.7,45,46 XRD identification of order/disorder is challenging because of overlapping peaks and interferences in kaolinite. The degree of structural order of affects modification strategies like mechanochemical activation and intercalation as discussed in Section 3.1 and 3.2. The degree of kaolinite XRD patterns exhibit broad band between 2θ = ∼12.2–35° with 4 peaks, which have basal spacing of about 0.71, 0.446, 0.358 and 0.256 nm, respectively.47 Hinckley index (HI) measures the degree of structural order in a kaolinite's crystal structure.48 Other techniques for analyzing kaolitic rocks consisting of not less than 20 wt% kaolinite include the weighting intensity ratio index (WIRI)49 and the Aparicio–Galan–Ferrell index (AGFI).50,52 HI values range from 0.3 to 1.8. Higher HI value indicates greater degree of structural order.51 For ordered kaolinites, the HI value is greater than 1.0 while HI is below 1.0 for disordered kaolinites.45,46 It is possible to obtain the HI value using the (02, 11) band which consists of the 02l and 11l sequences (20 to 23° 2θ CuK) because it is sensitive to random and interlayer displacements of type b/3 and 2.52 This index value is:
 | (1) |
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) reflection height above the band of the overlapping peak; B is the (11) reflection height above the band of the overlapping peak and Ah is the overall reflection height of the (10) peak above the background as shown in Fig. 2 below.47,52
0) reflection height above the band of the overlapping peak; B is the (11) reflection height above the band of the overlapping peak and Ah is the overall reflection height of the (10) peak above the background as shown in Fig. 2 below.47,52
 | ||
| Fig. 2 Fitting procedure for HI and AGFI parameter on (02, 11) band. | ||
Similarly, it is appropriate to obtain the AGFI value from the (02, 11) band. Conversely, weighting peak intensity ratio of (020), (10) and (11) peak reflection estimates AGFI value thus:
 | (2) |
For (10) and (11) peak intensities (IA and IB), the weighting coefficient is 1, while for (020) intensity, IC, the weighting coefficient, w = 2. Therefore, eqn (2) is rearranged thus:
 | (3) |
0) peak reflection intensity, IB is the (11) peak reflection intensity and IC is the (020) peak reflection intensity (Fig. 2). DIFPATAN (a computer program developed by Kuzel, 1991)53 computes the peak intensity values for IA, IB and IC appropriately.
WIRI is a function of the peak intensities of the (02, 11) band and full-width half maximum (FWHM) values of the corresponding (020), (10), (11) and (1) reflections resulting from decomposition of the same band. Hence, values of FWHM and corresponding intensities are obtainable from DIFPATAN.45
 | (4) |
0, (11), (1) and (020) peaks, respectively.54 Chmielova et al.45 modeled the relationship between HI and WIRI as HI = 0.10 + 1.44WIRI with R = 0.989. The relation: AGFI = 0.7–0.65WIRI + 1.86WIRI2 with R = 0.992 provides a model relationship between AGFI and WIRI. The value of WIRI ranges between 0 and 1. For low degrees of order, WIRI ≤ 0.4, for medium order, 0.4 < WIRI ≤ 0.7 and for highly ordered WIRI > 0.7.46
2.3. Surface charge heterogeneity
Hydrogen bonds bind together the tetrahedral sheets and the gibbsite-like sheets in the 1:1 layered silicate structure of kaolinite. Each bond is between an electronegative oxygen in the tetrahedral sheet and a hydroxyl group of the octahedral sheet in the c-axis.3 Consequently, kaolinite particles possess oppositely charged surface regions in aqueous media. One charge is permanently negatively charged on the T (tetrahedral) faces, which is extremely small. This arises from minor isomorphic substitution in the tetrahedral layer. The second is a variable charge on the O faces (hydroxyl-terminated planes) because of proton donation or acceptance of the edge groups.8,55,56 Depending on the pH of the protonation/deprotonation reaction in the aqueous phase, the charge on the amphoteric sites develops on the edges and faces of the octahedral (O) sheets. This causes charge heterogeneity.55,56 Protonation of aluminol groups occurs when kaolinite in aqueous medium possesses a pH below the point of zero net proton charge (PZNPC) of the main edge (amphoteric) site. Conversely, deprotonation of silanol and aluminol groups occurs when the pH of kaolinite in aqueous medium is greater than the PZNPC of the main edge (amphoteric) site. Both gibbsite planes and broken edges of aluminol (Al–OH) sites accept protons at pH values below the PZNPC of amphoteric sites (∼6.0 to 6.5). Therefore, increasing the pH above the PZNPC value results in the formation of negative charge on the gibbsite planes and broken edges, which leads to a decrease in zeta potential.8 This makes the aluminum (Al–OH) groups and broken edges reactive.
Conversely, silanol (Si–OH) groups on the broken edges are only reactive at pH values above PZNPC.56 However, the hydroxyl groups on the O faces are less reactive than those on the aluminol and the silanol on the broken edges.56 The following reactions occur at gibbsite planes and broken edges leading to the formation of positive charges:
| Al–OH + OH− → Al–O− | (5) |
Similarly, the following reactions occur at gibbsite planes and broken edges leading to the formation of negative charges:
| Si–OH + OH− → Si–O−. | (6) |
| Al–OH + H+ → Al–OH2+. | (7) |
2.4. Electrokinetic properties
The influence of aqueous phase properties on kaolinite particle electro-osmotic permeability and zeta potential is of vital importance. Determination of the shear plane electrical potential, ζ also known as zeta potential is essential to obtain the potential determining ions (pdi) and isoelectric point (iep). The knowledge of this properties provide a more active strategy for effecting electro-kinetic soil remediation processes. Vane and Zang57 reported that the kaolinite zeta potential ranged from +0.7 mV at pH = 2 to −54 mV at pH = 10 with 0.01 M KCl ionic strength (Table 3). Further, Alkan et al.58 studied the effect of pH, solid–liquid ratio, and concentration and type of electrolyte on the electrokinetic properties of kaolinite using the microelectrophoresis technique. The properties were tested by suspension of kaolinite in aqueous solutions of divalent heavy metal salts such as Pb(NO3)2, CuCl2 and CoCl2 and mono-, di-, and trivalent salts such as, KCl, NaCl, LiCl, NaCH3COO, NaNO3, MgCl3, BaCl2, CaCl2, Na2CO3, AlCl3, Na2SO4, Na3PO4FeCl3. The experimental findings of Alkan et al.58 and Vane and Zang57 are as follows:| pH that creates PZN | Maximum | Minimum | Ref. | ||
|---|---|---|---|---|---|
| pH | Zeta potential (mV) | pH | Zeta potential (mV) | ||
| a With 0.01 MKCL.b Georgia Kaolinite.c Steswhite kaolinite.d Lewiston, Montana.e Na kaolinite.f Low salt concentration.g 0.14 M NaCl. | |||||
| 2 < pH < 3b | 10 | −54a | 2 | 0.7a | 57 |
| 4.5 | 11 | −85 | 3 | ∼8 | 62a,b |
| 9.5 | −65 | 3 | ∼−3 | 62 | |
| <3c | 12 | −32 | 3 | Minute | 63 |
| 4 | 11 | −40 | 3.5 | ∼7 | 64 |
| 2.2d,e | 10 | −40 | 0 | 2.2 | 65 |
| 11 | −30 | 3.5 | 5.5 | 66 | |
| 6 | 12 | −40 | 2 | 10 | 67 |
| 7 and 11 | −25 | 3 | −8 | 68f,a | |
| 11 | −43 | 3 | −25 | 68g,b | |
(I) Kaolinite zeta potential and electro-osmotic permeability are strongly affected by pH, they both increase with increase in pH. This is in concomitant with the report of Wang et al.59
(II) The iep of kaolinite is at pH of about 2.35.
(III) Solid concentration does not significantly affect the zeta potential of kaolinite. The report of Kaya et al.,60 also agrees with this.
(IV) Monovalent cations and anions, and di- and trivalent anions are indifferent electrolytes for kaolinite whereas CuCl2, BaCl2, CaCl2, CoCl2, MgCl2, Pb(NO3)2, FeCl3 and AlCl3 altered the interface charge from negative to positive.
Further, the zeta potential of kaolinite is a function of its degree of structural order. A well-ordered kaolinite exhibits a lower zeta potential compared a poorly ordered one.61
2.5. Cation exchange capacity (CEC)
CEC quantifies the ability of kaolinite to exchange cations and retain nutrients. It informs the adsorption capacity of clay material because of its influences on the interaction with contaminants.69 When a contaminants interacts with kaolinite, the charge and the mechanical properties are affected. The CEC of a sample could be determine using an ammonia-specific electrode ammonia as the exchanged cation according to the procedure suggested by Worrall.70 The CEC of kaolinite depends on the pH and the amount of impurities it contains. Kaolinite material with high impurity or high pH possesses high CEC.71 In addition, the CEC of kaolinite is a function of its degree of structural order. A well-ordered kaolinite exhibits a lower CEC compared a poorly ordered one.722.6. Thermal properties of kaolinite
Comprehensive understanding of kaolinite thermal behavior is essential to deduce the results of thermal analysis. Thermal analysis gives information on weight loss, recrystallization, decomposition and phase transformation.73 Thermogravimetry/differential thermal analysis (TG/DTA) or differential scanning calorimetry (DSC) analyzes reveal the thermal behavior of the kaolinite structure.74 Kaolinites exhibit two endothermic peaks (at ∼55 to 80 °C and ∼450 to 600 °C) and one exothermic peak (at ∼980 to 1000 °C) (Fig. 3a). The first endothermic peak is of low intensity with an associated weight loss of ∼0.85 wt%, which is due to water physically absorbed from the atmosphere and held within particle defects. The second endothermic peak with higher intensity is because of a weight loss of ∼11.2 to 14.2 wt%.43 This is due to dehydroxylation of kaolinite particles arising from interactions between OH units released from the gibbsite-like sheets.20,74,75 Theoretically, the weight loss of the dehydroxylized kaolinite is 13.96 wt%.76 This highlights the importance of dehydroxylation as it results in the formation of metakaolin as shown in the reaction below:| Al2Si2O5(OH)4 → Al2O3·2SiO2 + 2H2O | (8) |
 | ||
| Fig. 3 TG/TDA of (a) kaolinite and (b) metakaolin.45 | ||
Dehydroxylation occurs simultaneously via homogeneous and heterogeneous mechanisms. Two neighboring OH-groups with different (less than and greater than the PZNPC) pH values interact in the homogeneous mechanism. The proton on the –OH unit with pH value less than the PZNPC reacts with one that has a pH value greater than PZNPC to release water through the interlayer spaces of the kaolinite particles. The increase in electrical conductivity at the dihydroxylation temperature74 during the heterogeneous mechanism induces the migration of protons produced in the kaolinite structure to the elimination region. This enhances the interaction with the OH units and subsequent water formation. Plausibly, these explain the variations in the peak temperature. The causes of the variations include degree of structural order, pressure, water vapor partial pressure, ultrasound processing, heating rate and degree of mechanochemical activation. A higher degree of structural order produces a higher endothermic peak while a smaller particle size leads to a lower endothermic peak.8,20,74
The exothermic transformation from the endothermic peak at ∼700 °C and subsequently, another exothermic peak appeared at ∼980 to 1000 °C. These are attributed to transformation of metakaolin to spinel or γ-alumina and amorphous silica. The latter transformation starts at ∼920 °C and lasts till ∼1100 °C. However, the precise product obtained from this transformation is still under investigation:76
| 2(Al2O3·2SiO2) → 2Al2O3·3SiO2 (spinel) + SiO2 | (9) |
| Al2O3·2SiO2 → Al2O3 (γ-alumina) + 2SiO2 | (10) |
A mullite phase starts to form at ∼1100 °C and increases with heating until 1300 °C. Subsequently, crystalline cristobalite emerges from the amorphous silica (eqn (12)).
| 3(2Al2O3·3SiO2) → 2(3Al2O3·2SiO2) (mullite) + 5SiO2 | (11) |
| SiO2 (amorphous) → SiO2 (cristobalite) | (12) |
3. Modification methods
As we alluded in Section 2, manipulating the physicochemical properties of catalytic materials is one of the surest means of enhancing activity and product selectivity. Hence, this section provides specific instances on different modification methods for improving kaolinite properties such as surface area, porosity, and acidity.3.1. Mechanochemical activation
This is a modification route that entails particle size reduction and helps to increase the pore volume and surface area of kaolinite materials. It culminates in improved surface reactivity as well as a higher capacity for ion exchange.18,77 Mechanochemical modification can be via specific micronization processes or industrial grinding.77 However, mechanochemical modification leads to delamination of kaolinite particles and agglomeration of the delaminated particles to form huge spheroidal particles. The delaminated product is a randomly structured wet xerogel in which the hydroxyl units react to form coordinated water molecules attached to the active sites on the surface of the modified kaolinite. This is because of water molecule formed from the interaction between the two –OH units (eqn (14)) by proton migration in a process called prototropy.18,19| OH− ↔ H+ + O2− | (13) |
| H+ + O H− ↔ H2O | (14) |
Expectedly, grinding decreases structurally ordered kaolinite that coexist with the disordered materials. This affects the degree of structural order in the delaminated precursor which determines the physicochemical properties of the mechanochemically activated kaolinite. The effect of mechanochemical activation of the kaolinite is greater for the kaolinite with high structural order compared to the kaolinite with low structural order.78 Moreover, excess grinding alters kaolinite crystal structure and turns it into an amorphous material with decreased surface area and pore volume. Torres et al.79 reported that delamination results in alumination of the kaolinite surface due to the formation of a coating of Al2O3 generated from the grinding process. This leads to enrichment in octahedral layer of the kaolinite and a corresponding increase in the PZNPC.75 Dellisanti et al.77 reported no appreciable change until after ∼5 h grinding at low spinning velocity which corresponds to 0.25 h at a high-speed spinning velocity. A bimodal particle size distribution appeared and increased appreciably with a great decrease in frequency volume (Fig. 4). This bimodal distribution after 5 h of grinding gradually transforms into a modal particle diameter after 10 to 20 h of grinding (Fig. 4). This is because of the agglomeration of delaminated particles to form huge spheroidal particles.16 Dellisanti et al.77 reported that mild grinding at low spinning velocity for 1 h does not affect the coherent scattering domain (CSD) and there is no accumulation of microstrain in the kaolinite structure. However, grinding at low spinning velocity (corresponding to 0.25 to 1 h at high-speed) for 5 to 10 h decreases the value of CSD with an associated increase in the kaolinite's microstrain structure. This is because of an increase in the FWHM value of the (001) diffraction peak resulting from a reduction in crystal size.17,77 Average crystal deformation could be represented by microstrain measured along the c-direction (001). Further grinding above 10 h increases the amorphousness of the kaolinite structure slightly but microstrain and CSD values remain constant.76 Grinding destroys the crystal structure of kaolinite by rupturing the O–H, Al–O–Si, Si–O, and Al–OH bonds80 Table 2 presents kaolinite FTIR results investigated by several authors. FTIR spectral changes occur after 5 to 10 of grinding (Fig. 4). These changes include peak broadening, reduction in peak intensity and the emergence of two broadband associated with water molecules absorbed by ground samples. These bands faded or likely disappeared with increasing in grinding time.77 Consequently, longer grinding time reduces surface structural variations. Further, it increases the number of acidic centers significantly. Interestingly however, further grinding of the super-active centers increases basicity.81,82
 | ||
| Fig. 4 Kaolinites particle size distribution before and after grinding.76 | ||
The study using mid-IR and near-IR spectroscopy also demonstrate the destruction in the kaolinite crystal structure by the rupturing of the O–H, Al–O–Si, Si–O, and Al–OH bonds. The study of Frost et al.83 followed this destruction using near-IR spectroscopy. The intensities of the two intense characteristic bands of kaolinite at 7065 and 7163 cm−1, which are in the spectral region of the first overtone of the hydroxyl-stretching vibration decreased upon mechanochemical activation. However, two new bands, which were assigned to the first overtone of the hydroxyl-stretching band of water emerged at 6842 and 6978 cm−1. Concurrently, mechanochemical activation increases the intensities of the water combination bands at 5238 and 5161 cm−1.
3.2. Thermal activation
Calcination in a temperature range of 550 to 950 °C modifies kaolinite into metakaolin. The aim of this modification route is to improve the thermal stability of the kaolinite material and prepare it for further modifications such as intercalation and chemical activation.23 Also, thermal activation breaks the hydrogen bonds between the dioctahedral layers in the 1:1 phyllosilicate material. This leads to evolution of the alumina coordinate towards AlVI–O, separation into aluminol-rich and silicol-rich domains and the subsequent formation of spinel phase.46,79
In the TG/DTA analysis of metakaolin exhibits only one peak. This is an exothermic peak at ∼980 to 1000 °C (Fig. 3b).46 However, no weight loss is associated with the exothermic peak. This is because of the germination of mullite and amorphous silica extraction from the metakaolin. The product of this synthesis is amorphous aluminosilicate which is more reactive than kaolinite. The metakaolin formed from thermal activation is a cementitious material used as a pozzolan18,47 and serves as efficient starting material for chemical lixiviation.23
Vizcayno et al.47 studied the XRD pattern of metakaolin and showed a pattern of reduced peak intensity in the 001 basal plane (2θ). This is due to the breakage of hydrogen bonds between the layers of kaolinite. This modification affects the basal plane peaks. Further, thermal activation at 700 °C leads to dehydroxylation of the entire moisture content of kaolinite. However, the moisture contents of the mica quartz in the solid material remain unaltered. FTIR spectra reveal the emergence of another band at ∼820 cm−1. This is because of the evolution of alumina coordinate towards AlVI–O. Furthermore, the Si–O–Al bending vibration vanishes because of deformation of the 1:1 dioctahedral layers. This alters the Si–O and Si–O–Si stretching vibrations at ∼1030, ∼1039, and ∼1113 cm−1.47,84,85
3.3. Intercalation
Intercalation is the effective insertion of a neutral polar organic substance into the interlamellar spaces of an inorganic host lattice in a regularly spaced stack to produce nano-sized composite materials. Wide interlamellar spaces prevent interactions between randomly dispersed layers in a consistent polymer matrix. This tends to facilitate exfoliation of the synthesized material. Kaolinite's unique, remarkably noncentrosymmetric structure is a suitable host for intercalation.86 Factors affecting intercalation include crystallinity and the particle size of the host material. Greater crystallinity induces higher rate of intercalation while smaller particle size causes lower intercalation rate. This is because the degree of structural order declines with reduced particle size.87,88 Intercalated kaolinite are efficient adsorbents,89 catalysts, catalyst supports and ion exchange agents.90 However, the main obstacle hindering kaolinite application as a host is the rigidity of its interlamellar space and lack of charge. Therefore, only a few guest compounds like methanol, acetamide, octadecyl amine, dimethylformamide, potassium acetate, N-dimethylformamide, deuterated dimethylsulfoxide and dimethylsulfoxide (DMSO) can intercalate directly into kaolinite layer. The most commonly used starting guest material is DMSO, because it possesses high thermal stability.90,91 However, the obtained intercalates after introducing guest compounds could undergo further intercalation of organic guests that cannot intercalate directly.81,92 Covalent grafting of small polar molecules such as DMSO or methanol achieves this modification. The guest compound migrates to the interlamellar aluminol basal plane of the kaolinite to generate Al–O–C bonds and expand the space. Subsequently, the second guest can then displace the first guest.93 Another intercalation method is co-grinding of solid organic guest and inorganic host. This method is called mechanochemical intercalation.81,75,94Several authors81,86,87 have studied kaolinite–DMSO intercalation XRD patterns. They reported the formation of kaolinite–DMSO intercalate results in a basal spacing increment of approximately 0.4 nm. The percentage DMSO intercalation is greater than 90%. This is possible because DMSO aggregates in aqueous solution and become more amorphous because of its large dipole moment. The presence of a large amount of free substratum under hydrothermal conditions results in a high rate of intercalation.90 Intercalating potassium acetate (KAc) into kaolinite with DSMO as the starting guest material results in ∼0.71 nm expansion. Furthermore, two small peaks emerge at ∼8.7 and ∼9.8 2θ. This is because DSMO deintercalates after drying under vacuum.95–97 Several FTIR spectra studies have shown the emergence of additional bands at 3663 and 905 cm−1, a reduction in the intensity of the 3696 cm−1 band with no changes to the 3622 cm−1 band. The 3663 cm−1 band is due to the vibration of inner surface hydroxyl stretching attached to the DMSO with hydrogen bonds. Conversely, the band at 905 cm−1 is due to the deformation of inner surface hydroxyl units attached to DMSO S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O units with hydrogen bonds. Vibration of the in-plane bending and out-plane bending of C–H bonds produces the 3023 and 2937 cm−1 bands. However, the 3669 and 3654 cm−1 bands disappear after intercalating DMSO into kaolinite. Studies have also shown subsequent disappearance of the 3663 cm−1 band and emergence of bands at 3698 and 3610 cm−1 when KAc is intercalated with kaolinite. Similarly, deintercalating DMSO in KAc solution leads to resurfacing of the pleochroic band at 3698 cm−1. Consequently, the presence of the 3610 cm−1 band shows the successful intercalation of KAc into the kaolinite structure.90,98 Table 4 presents the details of numerous FTIR assignments highlighting the effects of intercalation. Similarly, intercalating kaolinite with methanol causes space expansion of ∼0.37 to 0.4 nm. This expansion is the same as the molecular diameter of methanol. This indicates the presence of methanol and weak interaction between the methanol and kaolinite. However, the percentage intercalation of methanol is ∼93.7.98
O units with hydrogen bonds. Vibration of the in-plane bending and out-plane bending of C–H bonds produces the 3023 and 2937 cm−1 bands. However, the 3669 and 3654 cm−1 bands disappear after intercalating DMSO into kaolinite. Studies have also shown subsequent disappearance of the 3663 cm−1 band and emergence of bands at 3698 and 3610 cm−1 when KAc is intercalated with kaolinite. Similarly, deintercalating DMSO in KAc solution leads to resurfacing of the pleochroic band at 3698 cm−1. Consequently, the presence of the 3610 cm−1 band shows the successful intercalation of KAc into the kaolinite structure.90,98 Table 4 presents the details of numerous FTIR assignments highlighting the effects of intercalation. Similarly, intercalating kaolinite with methanol causes space expansion of ∼0.37 to 0.4 nm. This expansion is the same as the molecular diameter of methanol. This indicates the presence of methanol and weak interaction between the methanol and kaolinite. However, the percentage intercalation of methanol is ∼93.7.98
| Suggested assignments | Wave numbers (cm−1) | |||
|---|---|---|---|---|
| Crude-kaolinite | Kaolinite–DSMO | Kaolinite–Kac | Kaolinite–MeOH | |
| Inner surface –OH in-phase stretching (strong) vibration | 3696 | 3697 | 3698 | 3700 |
| Inner surface –OH out-phase stretching (medium) vibration | 3669 | 3663 | 3660 | |
| — | — | — | ||
| Inner surface –OH stretching (strong) vibration | 3622 | 3621 | 3610 | 3620 |
| 3540 | 3540 | |||
| 3500 | ||||
| In-plane bending of the vibration of C–H | 3023 | 3031 | ||
| Out-plane bending of the vibration of C–H | — | 2937 | — | 2969 |
| Physical absorbed water –OH vibration | 1629 | 1635 | 1640 | 1630 |
| Apical Si–O in-plane stretching vibration | 1107 | 1103 | 1107 | |
| Skeleton Si–O–Si in-plane stretching vibration | 1034 | 1032 | 1035 | 1031 |
| Al–OH bending vibration | 913 | 908 | 899 | 913 |
| AL–OH (“gibbsite-like” layer) translational vibration | 753 | 746 | 785 | 794 |
| 695 | 689 | 698 | 692 | |
| 539 | 544 | 555 | 689 | |
| Si–O–Al bending vibration | 470 | 467 | 478 | 469 |
| Si–O bending vibration | 431 | 433 | 438 | 430 |
Interestingly, intercalating 1-butyl-3-methylimidazolium bromine [brmim]Br into the methanol-pretreated kaolinite increases the space expansion to ∼0.7 nm.98,99 There have been several studies on thermal analysis of intercalates93,98,100 such as kaolinite–DMSO, kaolinite–polystyrene, and kaolinite–[bmim]Br using TG-DSC. The first weight loss similar to that of crude kaolinite appears at ∼80 °C. This is because of the loss of externally absorbed water. Subsequent partial deintercalation of DMSO occurs at 190 °C while dehydrolysis of kaolinite DMSO intercalate occurs at ∼479 °C.
The dehydroxylation temperature of crude kaolinite is slightly higher than that of kaolinite–DMSO. Similarly, there is partial deintercalation of 1-butyl-3 methylimidazolium bromine [brmim]Br at 277 °C followed by simultaneous decomposition of [brmim]Br and dehydroxylation of kaolinite at ∼470 and 545 °C respectively. This shows the presence of stronger hydrogen bond between the kaolinite hydroxyl layer and [brmim]Br than the bond between kaolinite layers. This culminates to increase in thermal stability of the kaolinite material.98 Ovramenko et al.101 showed that combined effect of intercalation and mechanochemical activation decreases the surface acidity of kaolinite. This is because longer grinding time destroys the structural OH groups and the kaolinite structural order. This decreases the degree of intercalation and forms super-active centers on the kaolinite surface.82 In situ decomposition of formamide bonded to these centers at 200 to 350 °C forms CO and NH3.82 Hence, the significant effect that intercalation has on the acid–base properties of kaolinite super-active centers.
3.4. Alkali activation
Alkali activation also modifies the acidity, surface area, pore size and volume as well as the adsorption strength of the kaolinite. This makes it a suitable precursor for solid basic catalyst.102 Alkali activation deprotonates aluminol and silanol groups from kaolinite materials. This leads to simultaneous dealumination and desilication of the kaolinite materials.8,103–106 Alkali activation is also a suitable method for developing a variety of basic zeolites with low Si/Al ratios. These include K–F zeolite, 13X zeolite,107 A, P and X zeolite,108,109 zeolite N,110 Na–Y zeolite,111,112 MCM-41 (ref. 113) and zeolite NaA.114 However, the extent of dealumination is usually low or insignificant in some instances.114,115 However, Kumar et al.116 reported that kaolinite treated with 3 M NaOH at 110 °C exhibits significant changes. These changes include 23 to 76 m2 g−1 surface area and 0.361 to 0.591 cm3 g−1 pore volume. Further, Si/Al ratio and acidity increased from 0.82 to 1.688 and 0.049 to 0.112 mmol g−1 respectively. In contrast, Belver et al.117 reported that kaolinite treated with 5 M KOH at 90 °C after calcination at 600 °C for 6 h exhibits adverse effects due to high molar concentration. The affected parameters were surface area from 18 to 4.1 m3 g−1, pore volume from 80 to 7.9 cm3 g−1, Si/Al ratio from 1.783 to 1.74 while acidity increased from 0.1049 to 0.1302 mmol g−1. However, 24 h treatment reduced the acidity to 0.0872 mmol g−1 due to neutralization of acid sites formed as a result of excessive leaching.Furthermore, acid activation followed by alkali activation is another technique for synthesizing ZSM-5 zeolite via template or template-free synthesis.118–121 It is important to note that at ∼1000 °C, alkali-activated kaolinite exhibit an endothermic peak while crude kaolinite exhibit exothermic peaks from ∼200 to 600 °C (Fig. 5)102,121,122 due to hydroxylation. The results obtained from XRD studies show a progressive decrease in the peak intensity of the kaolinite structure. Similarly, FTIR studies show extreme weakness in the structural hydroxyl vibration bands of kaolinite.116 The dehydroxylation is because of the deformation of some layers in the matrix of the kaolinite material. The foregoing discussion highlights the economic and structural importance of alkali activated kaolinite. The abundance of these raw materials has economical advantageous in production of zeolites compared to synthetic chemicals. Moreover, alkali-activated kaolinite has low Si/Al ratio which is a required feature for synthesizing low silica zeolites.122 Since the uses of zeolites in several branches of industry have been increasing, the production of zeolites by economical ways has gained great importance in material science.
 | ||
| Fig. 5 TG/DTA curves of the (a) raw kaolinite and (b) samples with composition 100 Kaol, 100 SiS and, 16 NaOH, and 22H2O and cured at 80 °C for 24 h.73 | ||
3.5. Acid activation
Acid-activated kaolinite are popular in adsorption and ion exchange. The activation enhances the acidity, surface area, pore size and volume, their catalytic properties of kaolinite as well as adsorption capacity.123 Acid activation leads to dealumination, removal of mineral impurities, disaggregation of kaolinite particles, and external layer dissolution. This changes the structure and chemical composition of the kaolinite materials.124,125 This makes it a suitable precursor for solid acid catalyst synthesis for petrochemical processes.22 Aside from increasing the porosity of the kaolinite, acid activation also enhances acid centers and the surface area. Further, kaolinite is suitable as an inorganic host for intercalation and exfoliation.124 Acid activation protonates aluminol (AlOH) groups using hydrogen ions from the aqueous acid medium. This leads to dealumination (eqn (15) and (16)) and increases the Si/Al ratio of the synthesis materials.8 Acid activation facilitates absorption due to increase in CEC, pore volume and surface area.123,126,127 This increases the amount of water physically absorbed by the kaolinite material. Conversely, activating with high concentration decreases the structural and coordinated water. This increases the endothermic peaks of the synthesized material. However, recrystallization and dehydroxylation temperature increases the endotherm peaks.25,113 The endotherms are higher in kaolinite with a high degree of structural order than in those with a low degree of order.124 The synthesized material is a mixture of inactivated kaolinite, amorphous and hydrous aluminosilicate as well as some partially protonated silica lamellae.106 The solubility of kaolinite varies from acid to acid, the ratio of kaolinite to acid, operating temperature, leaching period, and kaolinite particle size as well as the concentration of the acid. Kaolinite solubility increases with acid concentration and leaching period, but excessive leaching leads to a decrease in its surface area.23,25Activation with inorganic acids is more effective in generating new surface acid sites. It can also lead to collapse of the kaolinite's structure because of excessive leaching of the octahedral layer. Conversely, organic acids do not generate new acid sites as effectively as mineral acids. Meanwhile, they preserve the structure of the kaolinite because of their low activation power.
Table 5 presents a detailed comparison between organic and inorganic acid activation. It also shows the textural properties of some kaolinite samples before and after modification. Evidently, specific surface area and pore volume increase with increasing molarity of the activator and activation time. Panda et al.25 proposed that under the same conditions, the solubility of kaolinite in HCl is less than in H2SO4. Further, the order of kaolinite solubility and subsequent increase in surface area (Table 5) attributed to acid activation is: CH3COOH < H3PO4 < HCl < H2SO4 < HClO4 < HNO3.25,125,128 Eqn (15) and (16) express the dealumination process and its effect on the Al/Si stoichiometry of kaolinite in aqueous acidic medium.
| Kaolinite sample | Properties before activation | Activation | Molf (M) | Temp (°C) | Time (h) | Properties after activation | Ref | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Notation | Origin | SSAa | Pvolb | Si/Al | Acc | Calcd T (°C) | Cheme | SSAa | Pvolb | Si/Al | Acc | Acsg | ||||
| a SSA: BET specific surface area (m2 g−1).b Pvol: pore volume (cm3 g−1).c Ac: acidity (mmol g−1).d Calc: calcination temperature (°C).e Chem: chemical activation.f Mol: molarity (M).g Acs: acid sites (μmol g−1).h TPW: 12-Tungstophosphoric acid. | ||||||||||||||||
| K1 | Neimenggu, China | 19.4 | 0.1 | 1.1 | H3PO4 | 10 | 110 | 2 | 166 | 0.59 | 16.63 | 31 | ||||
| K1 | Neimenggu, China | 19.4 | 0.1 | 1.1 | H3PO4 | 5 | 110 | 2 | 45.3 | 0.17 | 1.76 | 31 | ||||
| K2 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 10 | 110 | 4 | 143 | 1.18 | 8.09 | 25 | ||||
| K2 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 5 | 110 | 4 | 107 | 0.967 | 1.63 | 25 | ||||
| K2 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 3 | 110 | 4 | 83 | 0.67 | 1.3 | 25 | ||||
| KGa-1b | Georgia | 9.8 | — | HCL | 2 | 80 | 14 | 34.2 | — | 124 | ||||||
| Kga-2 | Georgia | 20.5 | — | HCL | 2 | 80 | 14 | 45 | — | 124 | ||||||
| KGa-1b | Georgia | 3.8 | — | H2SO4 | 0.25 | 100 | 3 | 15.6 | — | 9 | ||||||
| K3 | Unknown | 9 | 0.08 | 950 | H2SO4 | 12 | 90 | 4 | 112 | 0.13 | 106 | 22 | ||||
| K3 | Unknown | 9 | 0.08 | 850 | H2SO4 | 12 | 90 | 20 | 288 | 0.22 | 114 | 22 | ||||
| K3 | Unknown | 9 | 0.08 | 850 | H2SO4 | 12 | 90 | 4 | 239 | 0.17 | 312 | 22 | ||||
| K3 | Unknown | 9 | 0.08 | 950 | H2SO4 | 12 | 90 | 20 | 110 | 0.13 | 87 | 22 | ||||
| K4 | China | — | 2.38 | NaOH | — | 200 | 4 | 326 | — | 107 | ||||||
| Kflint | Para-Brazil | 24 | — | 950 | H2SO4 | 4 | 90 | 1 | 406 | — | 237.7 | 17 | ||||
| Kflint | Para-Brazil | 24 | 950 | H2SO4 | 1 | 90 | 1 | 76 | 72 | 17 | ||||||
| Kflint | Para-Brazil | 24 | 850 | H2SO4 | 4 | 90 | 1 | 341 | 147.4 | 17 | ||||||
| Kflint | Para-Brazil | 24 | 850 | H2SO4 | 1 | 90 | 1 | 27 | 35.5 | 17 | ||||||
| K8 | Jiangsu, China | 41.1 | — | 2.00 | Washing | — | 41.1 | — | 2.00 | 26 | ||||||
| Kflint | Para-Brazil | — | — | 960 | HCl and 20% TPWh | 0.1 | 200 | 4 | — | — | 5.57 | 30 | ||||
| KC | South Africa | 960 | HCl and 20% TPWh | 0.1 | 200 | 4 | 1.06 | 30 | ||||||||
| KC | South Africa | 960 | HCl and 20% TPWh | 0.5 | 200 | 4 | 0.901 | 30 | ||||||||
| KGa-1b | Para-Brazil | 12 | — | 1.90 | 950 | H2SO4 | 4 | 90 | 1 | 138 | — | 163.4 | 33 | |||
| Kga-2 | Para-Brazil | 20 | — | 1.93 | 950 | H2SO4 | 4 | 90 | 1 | 65 | — | 147.4 | 33 | |||
| Kflint | Para-Brazil | 24 | — | 1.93 | 950 | H2SO4 | 4 | 90 | 1 | 406 | — | 237.7 | 33 | |||
| KC | South Africa | 9 | — | 1.96 | 950 | H2SO4 | 4 | 90 | 1 | 233 | — | 250.5 | 33 | |||
| K5 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 10 | 110 | 4 | 143 | 1.18 | 8.09 | 15 | ||||
| K5 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 5 | 110 | 4 | 107 | 0.967 | 1.63 | 15 | ||||
| K5 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 3 | 110 | 4 | 83 | 0.67 | 1.3 | 15 | ||||
| K5 | Kolkata, India | 23 | 0.361 | 0.65 | H2SO4 | 1 | 110 | 4 | 69 | 0.489 | 0.81 | 15 | ||||
| K6 | Beijing, China | — | 1.62 | NaOH | — | — | — | 364 | 0.47 | 0.57 | 132 | |||||
| K7 | Beijing, China | — | 1.3 | 850 | H2SO4 | — | 180 | 24 | 198 | 0.168 | 83 | 121 | ||||
| K5 | Kolkata, India | 23 | 0.361 | 0.82 | 0.049 | — | CH3COOH | 3 | 110 | 4 | 38 | 0.504 | 0.885 | 0.11 | 132 | |
| K5 | Kolkata, India | 23 | 0.361 | 0.82 | 0.049 | — | H3PO4 | 3 | 110 | 4 | 42 | 0.658 | 0.972 | 0.11 | 132 | |
| K5 | Kolkata, India | 23 | 0.361 | 0.82 | 0.049 | — | HCl | 3 | 110 | 4 | 78 | 1.083 | 1.144 | 0.23 | 132 | |
| K5 | Kolkata, India | 23 | 0.361 | 0.82 | 0.049 | — | HNO3 | 3 | 110 | 4 | 86 | 1.124 | 1.782 | 0.34 | 132 | |
| K5 | Kolkata, India | 23 | 0.361 | 0.82 | 0.049 | — | NaOH | 3 | 110 | 4 | 76 | 0.591 | 1.688 | 0.11 | 132 | |
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 600 | HCl | 6 | 90 | 6 | 219 | 0.19 | 117 | ||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 700 | HCl | 6 | 90 | 6 | 172 | 117 | |||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 800 | HCl | 6 | 90 | 6 | 209 | 0.19 | 117 | ||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 900 | HCl | 6 | 90 | 6 | 50.7 | 117 | |||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 700 | HCl | 6 | 90 | 24 | 21.9 | 0.14 | 117 | ||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 700 | KOH | 1 | 90 | 6 | 8.5 | 117 | |||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 600 | KOH | 1 | 90 | 6 | 9.2 | 117 | |||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 700 | KOH | 5 | 90 | 6 | 3.7 | 8.1 | 117 | ||||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 600 | KOH | 5 | 90 | 6 | 4.1 | 7.9 | 0.11 | 117 | |||
| K2μm | Navalacruz, Spain | 18.2 | 80 | 0.105 | 600 | KOH | 6 | 90 | 24 | 2.4 | 0.09 | 117 | ||||
| K10 | Leghorn, Italy | 17 | 2.02 | 600 | HCl | 2 | 80 | 4 | 185 | 16.67 | 131 | |||||
| K10 | Leghorn, Italy | 17 | 2.02 | 600 | HCl | 6 | 80 | 4 | 318 | 17.7 | 131 | |||||
| K10 | Leghorn, Italy | 17 | 2.02 | 600 | H2SO4 | 1 | 80 | 4 | 164 | 4.65 | 131 | |||||
| K10 | Leghorn, Italy | 17 | 2.02 | 600 | H2SO4 | 3 | 80 | 4 | 250 | 21.86 | 131 | |||||
| PBK-K | Unknown | 26.6 | — | 550 | H2SO4 | 4 | 100 | 0.75 | 70.4 | — | 125 | |||||
| PBK-K | Unknown | 26.6 | — | 550 | HNO3 | 4 | 100 | 0.75 | 101 | — | 125 | |||||
| PBK-K | Unknown | 26.6 | — | 550 | HClO4 | 4 | 100 | 0.75 | 79.7 | — | 125 | |||||
| KS 1 | Ukraina | 25 | — | 500 | HCl | 3 | 105 | 5 | 29.6 | — | 125 | |||||
| Kflint | Para-Brazil | 17 | — | 850 | H2SO4 | 4 | 130 | 0.25 | 187 | — | 4.32 | 130 | ||||
| Kflint | Para-Brazil | 17 | — | 850 | H2SO4 | 4 | 130 | 0.13 | 150 | — | 0.84 | 130 | ||||
| Kflint | Para-Brazil | 17 | — | 850 | H2SO4 | 4 | 130 | 0.25 | 285 | — | 1.44 | 130 | ||||
| Kflint | Para-Brazil | 17 | — | 850 | H2SO4 | 4 | 130 | 0.13 | 246 | — | 0.57 | 130 | ||||
| Kflint | Para-Brazil | 17 | 950 | H2SO5 | 4 | 130 | 0.25 | 130 | ||||||||
| Ka | Southwestern | 19 | 0.103 | Kaolinite–porphyrin intercalate | — | 150 | 315 | 10 | 0.068 | 85 | ||||||
Interestingly, both organic and inorganic acids can activate kaolinite chemically.110,128–130 Recently, synthesizing solid acid zeolites from kaolinite for the petrochemical industry has received great attention from numerous consortia.128–130 These interests highlight the importance of acid-activated kaolinite as a zeolite precursor for converting heavy molecules.129 This is because the process is cheaper and it produces zeolites with enhanced pore size and structure are more suitable than those of conventional zeolites.21 However, the process requires higher calcination at ∼550 to 950 °C for effective chemical activation and obtaining metakaolin from kaolinite. This is because of the presence of strong hydrogen bonds between its layers makes the material resistant to chemical attack.23,106
| 3H2SO4 + Al2Si2O5(OH)4 → Al2(SO4)3 + 2H4SiO4 + H2O | (15) |
| 3SO3 + Al2O3 → Al2(SO4)3 | (16) |
From XRD structural analyzes, several studies proved that all the peaks in acid-activated kaolinite correspond to the peaks of the original kaolinite material. However, acid activation reduces the kaolinite peak intensities because of the decrease in the degree of structural order. Conversely, mild acid activation increases the structural order because of an accompanying increase in crystal size which is due to decrease in the peak FWHM value. Expectedly, at higher acid concentrations, the peak in the XRD diffractograms becomes blurred. This indicates the low degree of structural order and amorphous nature of the synthesized material.22,25,124
Similarly, FTIR spectral analysis showed the inner OH stretching vibration at ∼3620 cm−1 has little or negligible weakness after mild acid activation. Thus, as the stronger the acid concentration, the weaker the band at 3620 cm−1. This shows that protonation increases with increasing acid concentration. It also reveals how rate of dealumination and dihydroxylation increases. However, all the bands show little or no weakness in the OH vibration bending region under mild acid activation. Increasing the acid to moderate concentrations of ∼5 M reduces the bands significantly. Subsequently, all bands disappear at higher concentrations of ∼10 M. This is because of the transformation of kaolinite from a crystalline phase to an amorphous phase due to the gibbsite-like layer deformation.22,25,124 Moreover, Lewis site-bonded pyridines and Brønsted acid site-bonded pyridines emerge near 1446 and 1595 cm−1 and at 1489, 1546 and 1633 cm−1 respectively. This is due to the weakness of the H bonds on the surface of pyridine molecules.33,131 Table 6 shows the XRD properties and FTIR assignments of several chemically activated kaolinite after thermal treatment.
| Kaolinite sample | Modification strategy | Mola (M) | Time (h) | Characterization | Application | SSAb (m2 g−1) | Reaction | Convc (wt%) | Liquid yield (wt%) | Seld (wt%) | Acse | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Notation | Origin | XRD | FTIR | Temp. (°C) | Time (h) | ||||||||||
| a M: molarity (M).b SSA: BET specific surface area (m2 g−1).c Conv: conversion.d Sel: selectivity.e Acs: acid sites (μmol g−1).f TPW: 12-tungstophosphoric acid activation.g Isobutene selectivity.h Debutilation selectivity. | |||||||||||||||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 1 | There is disappearance of the three well resolved peak between 20° and 24° (2θ) an emergence a broad band between 15° and 25° (2θ). This is due to the complete breakdown of the kaolinite crystalline structure and a structural water loss because of the transformation of the gibbsite-like Al2O3 into penta- and tetra-coordinated Al groups | Esterification of oleic acid | 408 | 160 | 4 | 98.9 | — | 237.7 | 17 | ||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 1 | Esterification of oleic acid | 341 | 130 | 4 | 84.2 | 237.7 | 17 | ||||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.25 | Esterification of oleic acid | 187 | 100 | 0.5 | 95.2 | 130 | |||||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.13 | Esterification of oleic acid | 150 | 100 | 0.5 | 56.4 | 130 | |||||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.25 | Esterification of oleic acid | 285 | 100 | 0.5 | 83.3 | 130 | |||||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.13 | Esterification of oleic acid | 246 | 100 | 0.5 | 13.5 | 130 | |||||
| K8 | Jiangsu, china | Washing | Cracking | 41.1 | 460 | 13.4 | — | — | 26 | ||||||
| Kflint | HCl and 20% TPWf | 0.1 | 4 | Metakaoline treated with 12-tungstophosphoric acid (HPW) shows peaks at 10.55°, 25.69° and 34.59° (2θ) confirming the presence of HPW in crystalline form | Metakaoline supported 12-tungstophosphoric acid (HPW) in aqueous medium possesses three typical absorptions at 1072 cm−1 (P–O), 961 cm−1 (WO) and 884 cm−1 (W–Oc–W) |
Esterification of oleic acid | 100 | 2 | 97.21 | — | 29 | ||||
| KGa-1b | Georgia | H2SO4 activation | 4 | 1 | All the kaolinite peaks are replaced by a broad band between 20° and 30° (2θ) that could be attributed to an SiO2 amorphous phase, and emergence of three peaks at 30°, 44° and 56° (2θ) showing a shape of type IIb | Exhibits Lewis site-bonded pyridine near at 1446 and 1595 cm−1 and Brönsted site-bonded pyridine at 1489 and 1546 cm−1 as well as 1633 cm−1 relating to the weak H bond to the pyridine molecules surface | Esterification of oleic acid | 138 | 160 | 4 | 94.8 | — | 163.4 | 32 | |
| Kga-2 | Georgia | H2SO4 activation | 4 | 1 | All the kaolinite peaks are replaced by a broad band between 20° and 30° (2θ) that could be attributed to an SiO2 amorphous phase, and emergence of three peaks at 30°, 44° and 56° (2θ) showing a shape of type IIa | Exhibits Lewis site-bonded pyridine near at 1446 and 1595 cm−1 and Brönsted site-bonded pyridine at 1489 and 1546 cm−1 as well as 1633 cm−1 relating to the weak H bond to the pyridine molecules surface | Esterification of oleic acid | 65 | 160 | 4 | 72 | — | 22.9 | 32 | |
| Kflint | Para-Brazil | H2SO4 activation | 4 | 1 | All the kaolinite peaks are replaced by a broad band between 20° and 30° (2θ) that could be attributed to an SiO2 amorphous phase, and emergence of three peaks at 30°, 44° and 56° (2θ) showing a shape of type IIa | Exhibits Lewis site-bonded pyridine near at 1446 and 1595 cm−1 and Brönsted site-bonded pyridine at 1489 and 1546 cm−1 as well as 1633 cm−1 relating to the weak H bond to the pyridine molecules surface | Esterification of oleic acid | 406 | 160 | 4 | 98.9 | — | 237.7 | 32 | |
| KC | South Africa | H2SO4 activation | 4 | 1 | All the kaolinite peaks are replaced by a broad band between 20° and 30° (2θ) that could be attributed to an SiO2 amorphous phase, and emergence of three peaks at 30°, 44° and 56° (2θ) showing a shape of type IIb | Exhibits Lewis site-bonded pyridine near at 1446 and 1595 cm−1 and Brönsted site-bonded pyridine at 1489 and 1546 cm−1 as well as 1633 cm−1 relating to the weak H bond to the pyridine molecules surface | Esterification of oleic acid | 335 | 160 | 4 | 98.8 | — | 250.5 | 32 | |
| K5 | Kolkata, India | No activation | 10 | 4 | Cracking of polypropylene | 143 | 450 | 0.33 | — | 89.5 | — | 15 | |||
| K6 | Beijing, China | NaOH activation | XRD pattern similar to that of zeolite Y | Shows similar bands assigned to zeolite Y | Cracking of heavy crude oil | 364 | 500 | — | — | 74.4 | 37.3 | 141 | |||
| K7 | Beijing, China | H2SO4 activation | 24 | All the characteristic peaks of ZSM-5 peaks were observed | All the bands assigned to ZSM-5 were observed | FCC naphtha aromatization | 198.3 | 550 | — | — | 73.93 | 83 | 121 | ||
| K9 | A1-Azraq, Jordan | HCl activation | 2 | Debutylation of 2-tert-butylphenol | 230 | 0.5 | 94 | 25h | 128 | ||||||
| K9 | A1-Azraq, Jordan | HCl activation | 1 | Debutylation of 2-tert-butylphenol | 230 | 0.5 | 97 | 50h | 128 | ||||||
| K9 | A1-Azraq, Jordan | HCl activation | 0.5 | Debutylation of 2-tert-butylphenol | 230 | 0.5 | 96 | 28h | 128 | ||||||
| K9 | A1-Azraq, Jordan | H3PO4 activation | 1 | Debutylation of 2-tert-butylphenol | 230 | 0.5 | 97 | 50h | 128 | ||||||
| K9 | A1-Azraq, Jordan | CH3COOH activation | 1 | Debutylation of 2-tert-butylphenol | 230 | 0.5 | 92 | 31h | 128 | ||||||
| K10 | Leghorn, Italy | HCl activation | 2 | 4 | 2-Propanol dehydratation | 185 | 120 | 26.1 | 132 | ||||||
| K10 | Leghorn, Italy | HCl activation | 6 | 4 | Exhibits Lewis site-bonded at 1445 and 1596 cm−1, Bronsted site-bonded pyridine at 1547 cm−1 and disappearance of all strong acid sites from the surface | 2-Propanol dehydratation | 318 | 120 | 40.8 | 132 | |||||
| K10 | Leghorn, Italy | H2SO4 activation | 1 | 4 | Exhibits Lewis site-bonded at 1454 and 1622 cm−1, Bronsted site-bonded pyridine at 1547 and 1638 cm−1, and weakly bonded hydrogen at 1597 cm−1 | 2-Propanol dehydration | 164 | 120 | 99.4 | 132 | |||||
| K10 | Leghorn, Italy | H2SO4 activation | 3 | 4 | Disappearance of all strong acid sites from the surface | 2-Propanol dehydratation | 250 | 120 | 55.8 | 131 | |||||
| PBK-K | H2SO4 activation | 4 | 0.75 | Friedel Crafts alkylation | 70.42 | 80 | 0.5 | 86.12 | — | — | 128 | ||||
| PBK-K | HNO3 activation | 4 | 0.75 | Friedel Crafts alkylation | 100.9 | 80 | 0.5 | 87 | — | — | 128 | ||||
| PBK-K | HClO4 activation | 4 | 0.75 | Friedel Crafts alkylation | 79.69 | 80 | 0.5 | 86.5 | — | — | 128 | ||||
| KS 1 | Ukraina | HCl activation | 3 | 5 | Patterns of the original kaolinite and its acid activated form were similar and indicating that there are no serious changes taking place in the clay structure due to mild acid activation | It exhibit the same characteristic peak with the original kaolinite due to the fact that the acid activation is so mild | Absorption 3-methoxybenzaldehyde | 24.95 | 60 | 3 | 80 | — | — | 129 | |
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.25 | There is disappearance of the peaks and emergence a broad band between 15° and 35° (2θ) attributed to the presence of amorphous SiO2 phase, and three intense peak at 25.5°, 37.9° and 48.2° (2θ) due to formation of anatase or rutile TiO2 usually found in as accessories to kaolins in the Capim river region | Exhibits absorption bands of pyridinium ion at 1533 and 1636 cm−1 due to the presence of Brønsted sites and the bands at 1448 and 1627 cm−1 attributed to Lewis sites-bonded pyridine | Esterification of oleic acid | 187 | 115 | 0.67 | 96.5 | — | 4.32 | 130 | |
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.13 | Esterification of oleic acid | 150 | 100 | 0.5 | 56.4 | — | — | 130 | |||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.25 | Esterification of oleic acid | 285 | 100 | 0.5 | 83.3 | — | 130 | ||||
| Kflint | Para-Brazil | H2SO4 activation | 4 | 0.13 | Esterification of oleic acid | 246 | 100 | 0.5 | 13.5 | — | — | 130 | |||
| Ka | Southwestern Brazil | Kaolinite–porphyrin intercalate | 315 | There is disappearance of 7.14 Å peak of the kaolinite on the kaolinite basal spacing and emergence of a peak at 10.6 Å of the basal spacing of the kaolinite–porphyrin intercalate | The OH band on the kaolinite at 3618 cm−1 is adjusted to 3620 cm−1 and there is emergence of C–H band at 2924 and 2848 cm−1 | Oxidation of cyclohexanone | 10 | 80 | 24 | 85 | 100 | — | 85 | ||
| K3 | 12 | 20 | Isomerization of 1-butene | 110 | 6.1 | 90.2g | 87 | 22 | |||||||
| K3 | Unknown | H2SO4 activation | 12 | 20 | Acid treatment of metakaolin that consists essentially of Al2O3·2SiO2 metakaolinite, led to very weak alteration in the XRPD pattern | Isomerization of 1-butene | 288 | — | — | 18 | 78.78g | 114 | 22 | ||
| K3 | Unknown | H2SO4 activation | 12 | 4 | Isomerization of 1-butene | 112 | — | — | 7.5 | |
89.3g | 106 | 22 | ||
4. Application of modified kaolinite
4.1. As catalyst support
Several attributes such as abundant availability, flexural and thermal stability, high capacity for enzymatic applications, resistance to leaching, and microbial activities have enabled kaolinite to remain relevant for over 13 decades. Further, kaolinite are eco-friendly, porous, possess small size disk (SSD) and preserve their energy and chemical functionality with prolong applications.These features made them suitable for several industrial utilizations, most especially as catalyst supports.34,132,133 Most importantly, the low cost of kaolinite has made it an economic replacement for other catalyst support in starting materials such as mullite (3Al2O3·2SiO2), alumina (Al2O3), and cordierite (2MgO·2Al2O3·5SiO2). The price of kaolinite is about 100 times lesser than that of alumina.132 Also, the sintering temperature of kaolinite (∼1250 °C) is less than that of alumina (∼1600 °C). Besides these, kaolinite possess greater flexural strength and lower density than alumina. Kaolinite can also be blended with other materials such as dolomite, natural hydroxyapatite (bones: Ca10(PO4)6(OH)2), calcite (CaCO3) or feldspar.132 Kaolinite is effective catalyst support for numerous reactions. These include zero valent Fe nanoparticles for the removal of aqueous Cu2+ and Co2+ ions32 and bimetallic iron/nickel nanoparticles for use in waste water treatment.11 Others include 12-tungstophosphoric for oleic acid esterification,30,134 Cu2+ and Co2+ for enhanced thermal strength,135 and nickel oxide for steam gasification of asphaltenes.27 In addition, they can support transition metals (Fe, Ni, Co and Cu) for carbon nanotubes and carbon sphere synthesis,135–137 lipase for effective esterification,34 chitosan, and maghemite (γ-Fe2O3).13 Kaolinites also support cobalt for synthesis of 2,3-dihydrofuran,138 Fe for heterogeneous Fenton-like reactions,10 cobalt phosphomolybdate for decolorization of azo-stuff12 and antimony doped with tin oxide (Sb–SnO2).30,139 Kaolinites have rendered immense support in catalysis in improving surface area, stability in inorganic solvents, thermal, mechanical and chemical strength, aggregation, and reactivity as well as in controlling the leaching of metals, metal oxide catalysts and biocatalysts.11
It is possible to use kaolinite as a support directly or after modification by thermal or treatment or chemical activation to increase the surface area. Impregnation, co-precipitation, chemical vapor deposition (CVD), deposition, and adsorption from solution disperse the primary catalyst onto the support surface. Several factors favor catalyst dispersion onto the support. These include the crystal size of the support material, a PZNPC value of the support of less than 8, and the high ion exchange capacity of the catalyst. Others are low catalyst concentration, lower sintering temperature and competing ions in the solution.140,141 Calcination enhances catalyst incorporation onto the support by transforming the support material from a crystalline phase to an amorphous phase and the catalyst becomes insoluble.31
4.2. As stand-alone catalyst
Modified kaolinite have proved to be efficient solid catalysts and catalyst supports in various reactions. These include cracking, esterification, aromatization, alkylation, adsorption, oxidation and isomerization. This improved conversion, yield and selectivity in several reactions, as shown in Table 6 is attributed to their robust enhancement in surface area, thermal stability and acidity. It is apparent from the table that under the same activation conditions, conversion increases with increasing specific surface area.125,130 However, dehydration of 2-propanol with H2SO4-treated kaolinite exhibited a contrary trend.131 This is probably because of the disappearance of all of the strong acid sites from the synthesized solid acid catalyst at higher acid concentrations, despite the overall higher surface area.Haden et al.139 used this procedure to synthesize 22.6% NaY zeolite with Si/Al ratio of 4.53. To improve the faujasite zeolite yield, Haden et al.140 proposed the application of deflocculating agent to slurries of hydrated kaolin with higher levels of solids to aid spray drying. The authors separated the resulting microspheres into two (10/90) fractions by weight prior to calcination and alkali treatment. The calcination temperatures employed for the 10 and 90 fractions were below and above the clay exotherms respectively. To obtain Y zeolite with improved resistance to attrition and higher yield of 52% within shorter crystallization time, the authors posit mixing the two fractions before treating with caustic soda solution. In a recent study,148 Atta et al. dealuminated metakaolin with inorganic acid in synthesizing X zeolite. This increased the Si/Al ratio to the expected value rather than using sodium silicate as reported by Haden et al.139
Interestingly, the authors obtained 57% faujasite zeolite comprised of 34% X zeolite and 23% Y zeolite. They attributed this to the similar synthesis conditions for both zeolites such as Si/Al ratio, pH value, aging, reaction temperature and time. This seems improbable because149 used 22 h aging period to achieve 52% Y zeolite while146 used 72 h. However, Chandrasekhar et al.149 gave the plausible explanation. They proposed that purity and crystallinity of Y zeolite increase with longer aging periods. Hence, Haden et al. would have generated a higher percentage of Y zeolite if they had increased the aging period.
Recently, several author had studied synthesis of hierarchical mesoporous SAPOs with mild acidity to solve the problem of rapid deactivation.153,159–161 Zhu et al.160 synthesized hierarchical SAPO-34 composed of decussate zeolite slice units by using raw kaolin as a special silica and alumina source. The improved reactivity and selectivity of olefin in conversion dimethyl ether to olefin reaction are credited to the distinctive hierarchical network. Cui et al.153 also reported the efficiency of hierarchical cross-like SAPO-34 developed through hydrothermal process using polyethylene glycol as the mesostructure directing agent. The catalyst performed excellently achieving up to 96% olefins selectivity, which was ascribed to ease of diffusion of both reactants and product through the well-connected pore channels.
5. Conclusion
Kaolinites are regaining their seemingly diminished popularity in several industrial processes, owing to the interesting physicochemical properties and other attributes. Prominent amongst the properties are surface charge heterogeneity and the degree of structural order which are usually measured by HI, AGFI or WIRI. Kaolinites are economical and environmentally benign starting materials for the synthesis of cheaper and eco-friendly catalysts, catalyst supports, ion exchange materials and adsorbents. These make them suitable supplements and in some instances, more effective substitutes for conventional materials, especially in catalysis. However, to appreciate the industrial applications of kaolinite, advances are required to enhance their properties for optimal use as highlighted in this review. Noteworthy is that mechanochemical activation improves the surface area and pore volume of kaolinite. This improves reactivity and ion exchange capacity in kaolinite materials. However, longer grinding reduces the degree of orderliness and the crystallinity of the material. Similarly, thermal activation weakens the hydrogen bonds that hold the kaolinite layers together to dehydroxylate the materials. This paves the way for effective activation. Conversely, chemical activation takes advantage of kaolinite' charge heterogeneity attributed to its amphoteric nature. This is possible with or without previous modifications such as mechanochemical and thermal activation. Acid activation improves the acidity and surface area of the catalytic material. This acidity enhancement is made possible via protonation of the AlOH groups which was facilitated by kaolinite surface charge heterogeneity.Intercalation is another modification method of keen interest. Researchers are employing this in synthesizing nano-sized composite catalytic materials. These have higher performance in catalysis because they offer shorter diffusion times when compared with microporous materials. Further, intercalation does not destroy kaolinite structure compared with other activation methods. The degree of intercalation is mainly determined by the degree of structural order of the starting kaolinite. High the degree of structural order engenders high the degree of intercalation. The foregoing highlights the suitability of kaolinite as viable starting materials of choice in catalysis for synthesis of stand-alone catalysts and catalyst support which are essential in processes such as FCC, olefins production and biodiesel production.
Acknowledgements
This study was carried out with the aid of a research grant from Fundamental Research Grant Scheme (FRGS) Grant (project no: FP031-2013A) under University of Malaya.References
- D. M. A. Melo, J. A. C. Ruiz, M. A. F. Melo, E. V. Sobrinho and M. Schmall, Microporous Mesoporous Mater., 2000, 345–349, 38 Search PubMed
.
- J. Madejova, Vib. Spectrosc., 2003, 1–10, 31 Search PubMed
- W. Yang and A. Zaoui, Appl. Clay Sci., 2013, 98–106, 80 Search PubMed
- D. L. Bish and R. B. Von Dreele, Clays Clay Miner., 1989, 37, 289–296 CAS
- K. Okada, A. Shimai, T. Takei, S. Hayashi, A. Yasumori and K. J. D. MacKenzie, Microporous Mesoporous Mater., 1998, 289–296, 21 Search PubMed
- C. D. Madhusoodana, Y. Kameshima, A. Nakajima, K. Okada, T. Kogure and K. J. D. MacKenzie, J. Colloid Interface Sci., 2006, 724–731, 297 Search PubMed
- G. Varga, Epitoanyag, 2007, 6–9, 59 Search PubMed
- P. Hu and H. Yang, Appl. Clay Sci., 2013, 58–65, 74 Search PubMed
- K. G. Bhattacharyya and S. S. Gupta, Desalination, 2011, 66–75, 272 Search PubMed
- N. Daud and B. Hameed, Desalination, 2011, 291–29, 269 Search PubMed
- X. Liu, Z. Chen, Z. Chen, M. Megharaj and R. Naidu, Chem. Eng. J., 2013, 764–771, 223 CrossRef
- Q. Zhuo, H. Ma, B. Wang and L. Gu, J. Hazard. Mater., 2007, 81–87, 142 Search PubMed
- H. Y. Zhu, R. Jiang and L. Xiao, Appl. Clay Sci., 2010, 522–526, 48 Search PubMed
- A. Vaccari, Catal. Today, 1998, 53–71, 41 Search PubMed
- A. K. Panda and R. Singh, J. Fuel Chem. Technol., 2011, 198–202, 39 Search PubMed
- A. Vaccari, Appl. Clay Sci., 1999, 161–198, 14 Search PubMed
- L. A. S. do Nascimento, L. M. Z. Tito, R. S. Angélica, C. E. F. da Costa, J. R. Zamian and G. N. R. Filho, Appl. Catal., B, 2011, 495–503, 101 Search PubMed
- E. Horvátha, R. L. Frost, É. Makóc, J. Kristófd and T. Csehd, Thermochim. Acta, 2003, 227–234, 404 Search PubMed
- É. Makó, Z. Senkár, J. Kristóf and V. Vágvölgyi, J. Colloid Interface Sci., 2006, 362–370, 294 Search PubMed
- V. Vágvölgyi, J. Kovács, E. Horváth, J. Kristóf and É. Makó, J. Colloid Interface Sci., 2008, 523–529, 317 Search PubMed
- P. Suitch and R. Young, Clays Clay Miner., 1983, 357, 31 Search PubMed
- M. Lenarda, L. Storaro, A. Talon, E. Moretti and P. Riello, J. Colloid Interface Sci., 2007, 537–543, 311 Search PubMed
- B. N. Dudkin, I. V. Loukhina, E. G. Avvakumov and V. P. Isupov, Chem. Sustainable Dev., 2004, 327–330, 12 Search PubMed
- M. Hart and D. Brown, J. Mol. Catal. A: Chem., 2004, 315–321, 212 Search PubMed
- K. P. Achyut, B. G. Mishra, D. K. Mishra and R. K. Singh, Colloids Surf., A, 2010, 98–104, 363 Search PubMed
- T. J. Rong and J. K. Xiao, Mater. Lett., 2002, 297–301, 57 Search PubMed
- A. Hassan, F. Lopez-Linares, N. N. Nassar, L. Carbognani-Arambarri and P. Pereira-Almao, Catal. Today, 2013, 112–118, 207 Search PubMed
- B. Nandi, A. Goswami and M. Purkait, J. Hazard. Mater., 2009, 387–395, 161 Search PubMed
- L. Shi, X. Zhang and Z. Chen, Water Res., 2011, 886–892, 45 Search PubMed
- O. S. Lacerda Júnior, R. M. Cavalcanti, T. M. de Matos, R. S. Angélica, G. N. R. Filho and I. C. L. Barros, Fuel, 2013, 604–611, 108 Search PubMed
- O. B. Ayodele, Appl. Clay Sci., 2013, 74–83, 72 Search PubMed
- Ç. Üzüm, T. Shahwana, A. E. Eroğlu, K. R. Hallam, T. B. Scott and I. Lieberwirth, Appl. Clay Sci., 2009, 172–181, 43 Search PubMed
- L. A. S. do Nascimentoa, R. S. Angélica, C. E. F. da Costa, J. R. Zamian and G. N. R. Filho, Appl. Clay Sci., 2011, 267–273, 51 Search PubMed
- M. B. Abdul Rahman, S. M. Tajudin, Z. Hussein, R. Z. R. Abdul Rahman, A. Salleh and M. Basri, Appl. Clay Sci., 2005, 111–116, 29 Search PubMed
- J. Huang, Y. Liu and X. Wang, J. Mol. Catal. B: Enzym., 2008, 49–54, 55 Search PubMed
- B. K. Speronello, Novel zeolite fluid cracking catalysts and preparation thereof from mixtures of calcined clay, US Pat., US4965233 A, 1990
- D. C. Bogert, L. B. Dight and M. A. Leskowicz, Ultra high zeolite content FCC catalysts and method for making same from microspheres composed of a mixture of calcined kaolin clays, US Pat., US5023220 A, 1991
- W. P. Hettinger Jr, Contribution to catalytic cracking in the petroleum industry, Appl. Clay Sci., 1991, 445–468, 5 Search PubMed
- Y. M. Sani and W. M. A. W. Daud, Appl. Catal., A, 2014, 140–161, 470 Search PubMed
- W. P. Gates, P. Komadel, J. Madejová, J. Bujdák, J. W. Stucki and R. J. Kirkpatrick, Appl. Clay Sci., 2000, 257–271, 16 Search PubMed
- M. J. Wilson, Clay mineralogy: spectroscopic and chemical determinative methods, ISBN 978-94-011-0727-3, Chapman & Hall, 1994, DOI:10.1007/978-94-011-0727-3
- M. Castellano, A. Turturro, P. Riani, T. Montanari, E. Finocchio, G. Ramis and G. Busca, Appl. Clay Sci., 2010, 446–454, 48 Search PubMed
- C. Hongfei, J. Yang, Q. Liuc, J. Zhang and R. L. Frost, Spectrochim. Acta, Part A, 2010, 856–861, 77 Search PubMed
- C. Belver, M. A. B. Munoz and M. A. Vicente, Chem. Mater., 2002, 2033–2043, 14 Search PubMed
- M. Chmielova and Z. Weiss, Appl. Clay Sci., 2002, 65–74, 22 Search PubMed
- K. L. Konana, C. Peyratouta, A. Smith, J.-P. Bonnet, S. Rossignola and S. Oyetola, J. Colloid Interface Sci., 2009, 103–109, 339 Search PubMed
- C. Vizcayno, R. M. de Gutiérrez, R. Castello, E. Rodriguez and C. E. Guerrero, Appl. Clay Sci., 2010, 405–413, 49 Search PubMed
- Y. Hu and X. Liu, Miner. Eng., 2003, 1279–1284, 16 Search PubMed
- A. Plancon and C. Zacharie, Clay Miner., 1990, 249–260, 25 Search PubMed
- P. Ptáček, D. Kubátová, J. Havlica, J. Brandštetr, F. Šoukal and T. Opravil, Thermochim. Acta, 2010, 24–29, 501 Search PubMed
- L. Stoch, Wydaw. geologiczne, Warszawa, 1974, p. 503 Search PubMed
- P. Aparicio and E. Galan, Clays Clay Miner., 1999, 12–27, 47 Search PubMed
- M. Valášková, M. Riedera, V. Matějka, P. Čapková and A. Slíva, Appl. Clay Sci., 2007, 108–118, 35 Search PubMed
- P. Ptáček, T. Opravil, F. Šoukal, J. Wasserbauer, J. Másilko and J. Baráček, J. Eur. Ceram. Soc., 2013, 2793–2799, 33 Search PubMed
- E. Tombacz and M. Szekeres, Appl. Clay Sci., 2006, 105–124, 34 Search PubMed
- P. V. Brady, R. T. Cygan and K. L. Nagy, J. Colloid Interface Sci., 1996, 356–364, 183 Search PubMed
- L. M. Vane and G. M. Zang, J. Hazard. Mater., 1997, 55, l–22 CrossRef
- M. Alkan, O. Demirbas and M. Dogan, Microporous Mesoporous Mater., 2005, 83, 51–59 CrossRef CAS
- J.-Y. Wang, X.-J. Huang, J. C. M. Kao and O. Stabnikova, J. Hazard. Mater., 2007, 144, 292–299 CrossRef CAS PubMed
- A. Kaya, A. H. Ören and Y. Yükselen, Mar. Georesour. Geotechnol., 2006, 24(3), 203–218 CrossRef CAS
- G. Tarı, I. Bobos, C. S. F. Gomes and J. M. F. Ferreira, J. Colloid Interface Sci., 1999, 210, 360–366 CrossRef PubMed
- Y. Hotta, T. Banno, Y. Nomura, S. Sano and K. Oda, J. Ceram. Soc. Jpn., 1999, 107(1249), 868–871 CrossRef CAS
- L. J. West and D. L. Stewart, Effect of zeta potential on soil electrokinesis, in The Proc. of Geoenvironment 2000, ASCE Special Publication, 1995, pp. 1535–1549 Search PubMed
- P. N. Lorenz, Clays Clay Miner., 1969, 17, 223–231 CAS
- R. W. Smith and Y. Narimatsu, Miner. Eng., 1993, 6, 753–763 CrossRef CAS
- D. J. A. Williams and K. P. Williams, J. Colloid Interface Sci., 1978, 65, 79–87 CrossRef CAS
- J. M. Dzenitis, Environ. Sci. Technol., 1997, 37, 1191–1197 CrossRef
- E. A. Stephan and G. G. Chase, Sep. Purif. Technol., 2001, 21, 219–226 CrossRef CAS
- K. H. Tan, Principles of Soil Chemistry, Marcel Dekker, New York, 1982 Search PubMed
- W. C. Worrall, Clays and Ceramic Raw Materials, Applied Science Pub., London, 1975, p. 203 Search PubMed
- J. C. Miranda-Trevino and C. A. Coles, Appl. Clay Sci., 2003, 23, 133–139 CrossRef CAS
- G. Tarı, I. Bobos, C. S. F. Gomes and J. M. F. Ferreira, J. Colloid Interface Sci., 1999, 210, 360–366 CrossRef PubMed
- F. Hussin, M. K. Aroua and W. M. A. W. Daud, Chem. Eng. J., 2011, 90–106, 170 Search PubMed
- H. Cheng, Q. Liu, J. Yang, S. Ma and R. L. Frost, Thermochim. Acta, 2012, 1–13, 545 Search PubMed
- E. Gasparini, S. C. Tarantino, P. Ghigna, M. P. Riccardi, E. I. Cedillo-González, C. Siligardi and M. Zema, Appl. Clay Sci., 2013, 417–425, 80 Search PubMed
- A. Ghorbel, M. Fourati and J. Bouaziz, Mater. Chem. Phys., 2008, 876–885, 112 Search PubMed
- F. Dellisanti and G. Valdre, Int. J. Miner. Process., 2012, 69–77, 102 Search PubMed
- G. Tarı, I. Bobos, C. S. F. Gomes and J. M. F. Ferreira, J. Colloid Interface Sci., 1999, 210, 360–366 CrossRef PubMed
- S. R. Torres, E. Basaldella and J. Marco, J. Colloid Interface Sci., 1999, 339–344, 215 Search PubMed
- E. Mako, R. L. Frost, J. Kristof and E. Horvath, J. Colloid Interface Sci., 2001, 244, 359–364 CrossRef CAS
- D. Sun, B. Li, Y. Li, C. Yu, B. Zhang and H. Fei, Mater. Res. Bull., 2011, 101–104, 46 Search PubMed
- E. Horvath, J. Kristof, R. L. Frost, E. Mako, E. Jakab and A. Redey, J. Colloid Interface Sci., 2005, 132–138, 289 Search PubMed
- R. L. Frost, É. Mako, J. Kristo and J. T. Kloprogge, Spectrochim. Acta, Part A, 2002, 58, 2849–2859 CrossRef CAS
- A. Tironi, M. A. Trezza, E. F. Irassar and A. N. Scian, Procedia Mater. Sci., 2012, 343–350, 1 Search PubMed
- N. Bizaia, E. H. de Faria, G. P. Ricci, P. S. Calefi, E. J. Nassar, K. A. D. F. Castro, S. Nakagaki, K. J. Ciuffi, R. Trujillano, M. A. Vicente, A. Gil and S. A. Korili, ACS Appl. Mater. Interfaces, 2009, 2667–2678, 1 Search PubMed
- T. A. Elbokl and C. Detellier, J. Phys. Chem. Solids, 2006, 950–955, 67 Search PubMed
- K. Song, X. Wang, P. Qian, C. Zhang and Q. Zhang, Comput. Theor. Chem., 2013, 72–80, 1020 Search PubMed
- Y. Deng, G. N. White and J. B. Dixon, J. Colloid Interface Sci., 2002, 379–393, 250 Search PubMed
- R. R. Da Silva and D. L. Guerra, J. Chil. Chem. Soc., 2013, 1678–1683, 58 Search PubMed
- P. C. Lopes, F. A. Dias and L. da Silva, Mater. Lett., 2003, 3397–3401, 57 Search PubMed
- J. Matusik, Z. Kłapyta and Z. Olejniczak, Appl. Clay Sci., 2013, 426–432, 83 Search PubMed
- J. Matusik and Z. Kłapyta, Science, 2013, 433–440, 83 Search PubMed
- S. Letaief, I. K. Tonle, T. Diaco and C. Detellier, Appl. Clay Sci., 2008, 95–101, 42 Search PubMed
- É. Makó, J. Kristóf, E. Horváth and V. Vágvölgyi, Appl. Clay Sci., 2013, 24–31, 83 Search PubMed
- B. Zhang, Y. Li, X. Pan, X. Jia and X. Wang, J. Phys. Chem. Solids, 2007, 135–142, 68 Search PubMed
- C. Johnston and D. Stone, Clays Clay Miner., 1990, 121–128, 38 Search PubMed
- H. Cheng, Q. Liu, J. Yang, X. Du and R. L. Frost, Appl. Clay Sci., 2010, 476–480, 50 Search PubMed
- L. Zhang, C. Wang, Z. Yan, X. Wu, Y. Wang, D. Meng and H. Xie, Appl. Clay Sci., 2013, 106–110, 86 Search PubMed
- S. Letaief and C. Detellier, J. Mater. Chem., 2007, 1476–1484, 17 Search PubMed
- T. A. Elbokl and C. Detellier, J. Colloid Interface
Sci., 2008, 338–348, 323 Search PubMed
- N. A. Ovramenko, O. F. Zakharchenko and F. D. Ovcharenko, Kolloidnyj Zhurnal, 1994, 410–12, 56 Search PubMed
- F. Slaty, H. Khoury, J. Wastiels and H. Rahier, Appl. Clay Sci., 2013, 120–125, 75–76 Search PubMed
- A. M. Rashad, Construct. Build. Mater., 2013, 751–765, 41 Search PubMed
- A. M. Rashad, Construct. Build. Mater., 2013, 303–318, 41 Search PubMed
- A. Poulesquen, F. Frizon and D. Lambertin, Rheological Behavior of Alkali-Activated Metakaolin During Geopolymerization, Springer, 2013, pp. 225–238 Search PubMed
- Q. Tan, X. Bao, T. Song, Y. Fan, G. Shi, B. Shen, C. Liu and X. Gao, J. Catal., 2007, 69–79, 251 Search PubMed
- Y. Ma, C. Yan, A. Alshameri, X. Qiu, C. Zhou and D. Li, Adv. Powder Technol., 2014, 495–499, 25 Search PubMed
- C. Belviso, F. Cavalcante, A. Lettino and S. Fiore, Appl. Clay Sci., 2013, 162–168, 80 Search PubMed
- M. Murat, A. Amokrane, J. P. Bastide and L. Montanaro, Clay Miner., 1992, 27, 119–130 CAS
- I. D. Mackinnon, G. J. Millar and W. Stolz, Appl. Clay Sci., 2012, 1–7, 58 Search PubMed
- D. M. EL-Mekkawi and M. M. Selim, J. Environ. Chem. Eng., 2014, 723–730, 2 Search PubMed
- P. A. Alaba, Y. M. Sani and W. M. A. W. Daud, Chin. J. Catal., 2015, 36, 1846–1851 CrossRef
- E. Du, S. Yu, L. Zuo, J. Zhang, X. Huang and Y. Wang, Appl. Clay Sci., 2011, 94–101, 51 Search PubMed
- A. R. Loiola, J. C. R. A. Andrade, J. M. Sasaki and L. R. D. da Silva, J. Colloid Interface Sci., 2012, 34–39, 367 Search PubMed
- D. Serrano and P. Pizarro, Chem. Soc. Rev., 2013, 4004–4035, 42 Search PubMed
- S. Kumar, A. K. Panda and R. Singh, Bull. Chem. React. Eng. Catal., 2013, 61–69, 8 Search PubMed
- C. Belver, M. A. B. Munoz and M. A. Vicente, Chem. Mater., 2002, 2033–2043, 14 Search PubMed
- P. A. Alaba, Y. M. Sani, I. Y. Mohammed, Y. A. Abakr and W. M. A. W. Daud, J. Taiwan Inst. Chem. Eng., 2015, 1–8, DOI:10.1016/j.jtice.2015.09.006
- F. Pan, X. Lu, Y. Wang, S. Chen, T. Wang and Y. Yan, Microporous Mesoporous Mater., 2014, 134–140, 184 Search PubMed
- F. Pan, X. Lu, Y. Wang, S. Chen, T. Wang and Y. Yan, Mater. Lett., 2014, 5–8, 115 Search PubMed
- P. Wang, B. Shen, D. Shen, T. Peng and J. Gao, Catal. Commun., 2007, 1452–1456, 8 Search PubMed
- M. Alkan, Ç. Hopa, Z. Yilmaz and H. Güler, Microporous Mesoporous Mater., 2005, 176–184, 86 Search PubMed
- K. G. Bhattacharyya and S. S. Gupta, Colloids Surf., A, 2006, 277, 191–200 CrossRef CAS
- M. Valášková, K. Barabaszová, M. Hundáková, M. Ritz and E. Plevová, Appl. Clay Sci., 2011, 70–76, 54 Search PubMed
- K. R. Sabu, R. Sukumar, R. Rekha and M. Lalithambik, Catal. Today, 1999, 321–326, 49 Search PubMed
- K. G. Bhattacharyya and S. S. Gupta, Ind. Eng. Chem. Res., 2007, 46, 3734–3742 CrossRef CAS
- K. G. Bhattacharyya and S. S. Gupta, Chem. Eng. J., 2008, 136, 1–13 CrossRef CAS
- S. Mahmoud and S. Saleh, Clays Clay Miner., 1999, 481–486, 47 Search PubMed
- B. Shen, P. Wang, Z. Yi, W. Zhang, X. Tong, Y. Liu, Q. Guo, J. Gao and C. Xu, Energy Fuels, 2009, 60–64, 23 Search PubMed
- A. D. N. de Oliveira, L. R. D. S. Costa, L. H. D. O. Pires, L. A. S. do Nascimento, R. S. Angélica, C. E. F. da Costa, J. R. Zamian and G. N. D. R. Filho, Fuel, 2012, 626–631, 103 Search PubMed
- M. Perissinotto, M. Lenarda, L. Storaro and R. Ganzerla, J. Mol. Catal. A: Chem., 1997, 103–109, 121 Search PubMed
- Y. F. Chang, S. N. Vaughn, L. R. M. Martens, J. W. Sprinkle and I. N. Walker, Molecular sieve catalyst composition, its making and use in conversion processes, WO2004060559 A1, 2004
- E. H. de Faria, G. P. Ricci, L. Marcal, E. J. Nassar, M. A. Vicente, R. Trujillano, A. Gil, S. A. Korili, K. J. Ciuffi and P. S. Calefi, Catal. Today, 2012, 187, 135–149 CrossRef CAS
- Y.-F. Chang, S. N. Vaughn, K. R. Clem, L. R. Martens and W. Hu, Molecular sieve catalyst composition, its making and use in conversion processes, US Pat., US7241713 B2, 2007
- Y. F. Chang, Molecular sieve catalyst composition, its making and use in conversion processes, WO2006083423 A1, 2006
- A. Kovo, O. Hernandez and S. Holmes, J. Mater. Chem., 2009, 6207–6212, 19 Search PubMed
- F. J. Dzierzanowski and W. L. Haden Jr, US Pat., US3433587 A, 1969
- W. L. Haden Jr and F. J. Dzierzanowski, Zeolitic catalyst and preparation, US Pat., 3,663,165, 1972
- W. L. Haden, Microspherical zeolitic molecular sieve composite catalyst and preparation thereof, US Pat., US3506594 A, 1970
- W. L. Haden, Microspherical zeolitic molecular sieve composite catalyst and preparation thereof, US Pat., US3647718 A, 1972
- M. C. D Jose and R. de Janeiro, US Pat., 5,082,815, 1992
- S. C. S. Joseph, E. W. Albers and G. R. Wilson, US Pat., 5,739,072, 1998
- A. P. L. Robert and K. A. Jocelyn, US Pat., 5,366,948, 1994
- S. K. Barry, US Pat., 4,628,042, 1986
- A. P. L. Robert and J. A. Herbst, US Pat., 5,231,064, 1993
- S. M. Brown and G. M. Woltermann, US Pat., US4157375 A, 1979
- S. M. Brown and G. M. Woltermann, US Pat., US4235753 A, 1980
- A. Atta, O. Ajayi and S. Adefila, J. Appl. Sci. Res., 2007, 1017–1021, 3 Search PubMed
- S. Chandrasekhar and P. Pramada, Appl. Clay Sci., 2004, 187–198, 27 Search PubMed
- W. Pengfei, L. Ailing, H. Jie, X. Jing'an and L. Guanzhong, Ind. Eng. Chem. Res., 2011, 9989–9997, 50 Search PubMed
- H. Q. Zhou, W. Yao, F. Wei, D. Z. Wang and Z. W. Wang, Appl. Catal., A, 2008, 112, 341 Search PubMed
- F. Niloufar, S. Morteza, S. J. Royaee and S. Mirarefin, Chem. Eng. Res. Des., 2011, 811–816, 89 Search PubMed
- Y. Cui, Q. Zhang, J. He, Y. Wang and F. Wei, Particuology, 2013, 468– 474, 11 Search PubMed
- T. N. Ali, F. Shohreh, S. Morteza and S. Maede, J. Ind. Eng. Chem., 2012, 29–37, 18 Search PubMed
- Z. M. Yan, B. H. Chen and Y. Huang, Solid State Nucl. Magn. Reson., 2009, 49, 35 Search PubMed
- H. O. Pastore, G. A. V. Martins, M. Strauss, L. G. Pedroni, G. B. Superti, E. C. de Oliveira, G. Gatti and L. Marchese, Microporous Mesoporous Mater., 2008, 81, 107 Search PubMed
- H. O. Pastore, E. C. de Oliveira, G. B. Superti, G. Gatti and L. Marchese, J. Phys. Chem. C, 2007, 3116, 111 Search PubMed
- T. Wang, L. Xuchen and Y. Yan, Microporous Mesoporous Mater., 2010, 138–147, 136 Search PubMed
- Y. Seung-Tae, K. Ji-Ye, C. Ho-Jeong, K. Min, J. Soon-Yong and A. Wha-Seung, Mater. Res. Bull., 2012, 3888–3892, 47 Search PubMed
- J. Zhu, Y. Cui, Y. Wang and F. Wei, Chem. Commun., 2009, 3282–3284 RSC
- L. Chen, R. W. Wang, S. Ding, B. B. Liu, H. Xia and Z. T. Zhang, et al., Chemical Journal of Chinese Universities, 2010, 1693–1696, 31 CrossRef
| This journal is © The Royal Society of Chemistry 2015 |