
Synthesis of degradable multi-segmented polymers via Michael-addition thiol–ene step-growth polymerization†
Joke
Vandenbergh
a,
Gijs
Ramakers
a,
Luk
van Lokeren
b,
Guy
van Assche
b and
Tanja
Junkers
*ac
aPolymer Reaction Design Group, Institute for Materials Research (imo-imomec), Hasselt University, Campus Diepenbeek, Building D, B-3590 Diepenbeek, Belgium. E-mail: tanja.junkers@uhasselt.be; Fax: +32 11 26 83 01; Tel: +32 11 26 83 18
bPhysical Chemistry and Polymer Science, Department of Materials and Chemistry, Vrije Universiteit Brussel, Pleinlaan 2, B-1050 Brussels, Belgium
cIMEC Associated Lab IMOMEC, Wetenschapspark 1, B-3590 Diepenbeek, Belgium
First published on 21st September 2015
Abstract
A series of (bio)degradable multi-segmented poly(β-thioester) (PBT) linear polymers and networks are synthesized using bifunctional telechelic polystyrene (PS) and poly(iso-bornylacrylate) (PiBoA) precursor polymers, obtained via reversible addition fragmentation chain transfer polymerization. The thiocarbonyl thio end groups of the bifunctional RAFT precursors are converted into thiols via aminolysis with hexylamine. The obtained dithiol polymers are then used as Michael donors in a phosphine catalyzed thiol–ene step-growth polymerization, either with hexanediol diacrylate to yield linear polymers in which the diacrylate units are evenly spaced along the backbone, or with multifunctional acrylates to obtain cross-linked PBT networks with defined PS segment sizes. Aminolysis and thiol–ene Michael addition reactions can be performed in a one-pot procedure, but improved results are obtained when the polymeric thiols are first purified. The multi-segmented poly(β-thioester) polymers based on polystyrene precursors are characterized by means of TGA and DSC. The materials are thermally very stable and feature glass transition temperatures between polystyrene and pure PBTs. Further, the (bio)degradability of the materials (into the original low molecular weight PS segments) is demonstrated by basic or enzymatic hydrolysis of the labile ester bonds in the polymer backbone.
Introduction
An important aspect in modern polymer science comprises the development of biodegradable polymer materials, targeting ecological applications such as recyclable low-cost commodity plastics for waste reduction or biomedical applications in controlled drug release, tissue engineering and medical implants.1,2 (Bio)degradable materials have the ability to disintegrate into smaller molecules upon interaction with a specific biological or physical trigger such as micro-organisms, enzymes, pH, temperature or light. Natural biodegradable polymers, for example poly(saccharides) and poly(peptides), are found in animals and plants. They excel in biocompatibility and are used in pharmaceuticals, foods, biofuels and many more applications.3–5 However their use for technological applications is to some extent limited due to batch-to-batch variations, improper mechanical properties or the lack of built-in specific functionalities. These drawbacks can be overcome by versatile synthetic polymers, which due to the vast variety of available monomers and synthetic polymerization pathways can be designed with distinct properties and functionalities and are thus fine-tuned for specific applications. The most well-known synthetic biodegradable polymers are poly(lactides) and poly(glycolides), which find applications in bulk materials, biomedical and ecological areas.6,7 Mostly, synthetic biodegradable polymers are synthesized via polycondensation or ring opening polymerization,8 but also other synthetic pathways are nowadays developed.9A possible synthesis methodology to obtain biodegradable polymers, is to make use of thiol–ene step growth polymerization.10 Thiol–ene chemistry has been studied extensively as route to produce (cross-linked) polymer materials for various applications.11 Homogeneous networks with excellent thermal, physical and mechanical properties can be produced by thiol–ene chemistry.12,13 The reaction can proceed either via a radical addition of a thiol to an ene-compound initiated by heat or light triggers,14–18 or via a base/nucleophilic catalyzed Michael addition of the thiol onto an electron deficient ene-compound such as acrylates, acrylamides or maleimides.19–21 Primary amines or phosphines are used as nucleophilic bases for the Michael addition reaction.22,23 Although the radical thiol–ene route is described most extensively in literature, the resulting polymers are not always degradable and the reaction sometimes suffers from formation of side products and occasionally from unsatisfactory yields.24 The Michael-type thiol–ene step-growth reaction on the other hand, features better orthogonality, is more reactive and allows in step-growth reactions for polymers with an overall higher degree of polymerization. As such, the reaction is often described as a click reaction.25,26 Furthermore, when acrylate monomers are used as electron deficient ene-compounds, the resulting polymers are prone to (bio)degradation since they carry hydrolysable ester bonds in their backbone.
Recently, we reported on the synthesis of a variety of biodegradable poly(β-thioester) (PBT) materials using Michael-type thiol–ene step-growth polymerizations.27–29 A library of both linear polymers as well as hyperbranched polymers and networks were synthesized from commercially available multifunctional thiol and acrylate monomers. The resulting PBT polymers contain labile ester bonds in their main chain and are hence prone to degradation. Degradability was demonstrated by means of hydrolysis studies in buffer solutions and by time-dependent weight loss measurements using quartz crystal microbalance (QCM) investigations.
Despite their high versatility, PBTs feature the disadvantage that dithiols are used in their synthesis. These compounds are often associated with strong odors. Even though toxicity of the monomers plays no significant role, this is an issue to address in order to make PBT materials more widespread. Also, most dithiols (or multifunctional thiols) contain alkyl or ethylene glycol chain segments, but in most cases no usable further chemical functionalities. A greater choice of polymerizable substrates is hence desirable. In the current study, the synthesis of PBT polymers was thus extended by using thiol homotelechelic polystyrene (PS) and poly(iso-bornyl acrylate) (PiBoA) precursor polymers as building blocks for PBT synthesis. In this fashion, the variety in chemical and mechanical properties of vinyl polymers is introduced into the realm of PBTs by combining the properties of the classical vinyl polymers with the properties of PBTs. In principle, such reactions open pathways towards degradable conventional vinyl polymers by forming multi-segmented polymers which are bonded by degradable moieties. Reversible addition fragmentation chain transfer (RAFT) polymerization30 was applied to synthesize the precursor polymers by using a custom-made bifunctional RAFT agent, bis-2-(dodecylthiocarbonothioylthio)propionic acid (BiDoPAT). The resulting polymers exhibit narrow molar mass distributions and high end group fidelities. After polymerization, the thiocarbonyl thio end groups of the resulting polymers were converted into thiols via aminolysis, a popular RAFT end group modification reaction.31,32 In the first subsequent experiments, the dithiol polymers were reacted in situ with diacrylates via a one-pot procedure. However, in optimized experiments, the dithiol precursor polymers were first purified to remove undesirable excess hexylamine and side products, and subsequently polymerized in a step-growth fashion with a diacrylate via phosphine catalyzed thiol–ene conjugations (Fig. 1). When simple diacrylates are employed as co-monomer, multi-segmented linear polymers are obtained which periodically contain the acrylate moieties. Next to degradability of such materials, this chemistry also opens novel pathways towards more complex polymer structures when functional acrylates were to be used. The synthesis of materials where certain functional groups are not randomly distributed, but evenly spaced along the polymer main chain is to date very challenging. Besides linear thiol–ene polymers, also multifunctional acrylate monomers were copolymerized to obtain high-molecular weight polymer networks. Those materials were further subjected to thermal characterization by means of in-depth differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Finally, the (bio)degradability of the build-in acrylate bridges between the polymer segments was demonstrated by hydrolyzing the ester bonds in the polymer backbone using basic conditions or porcine liver esterase.
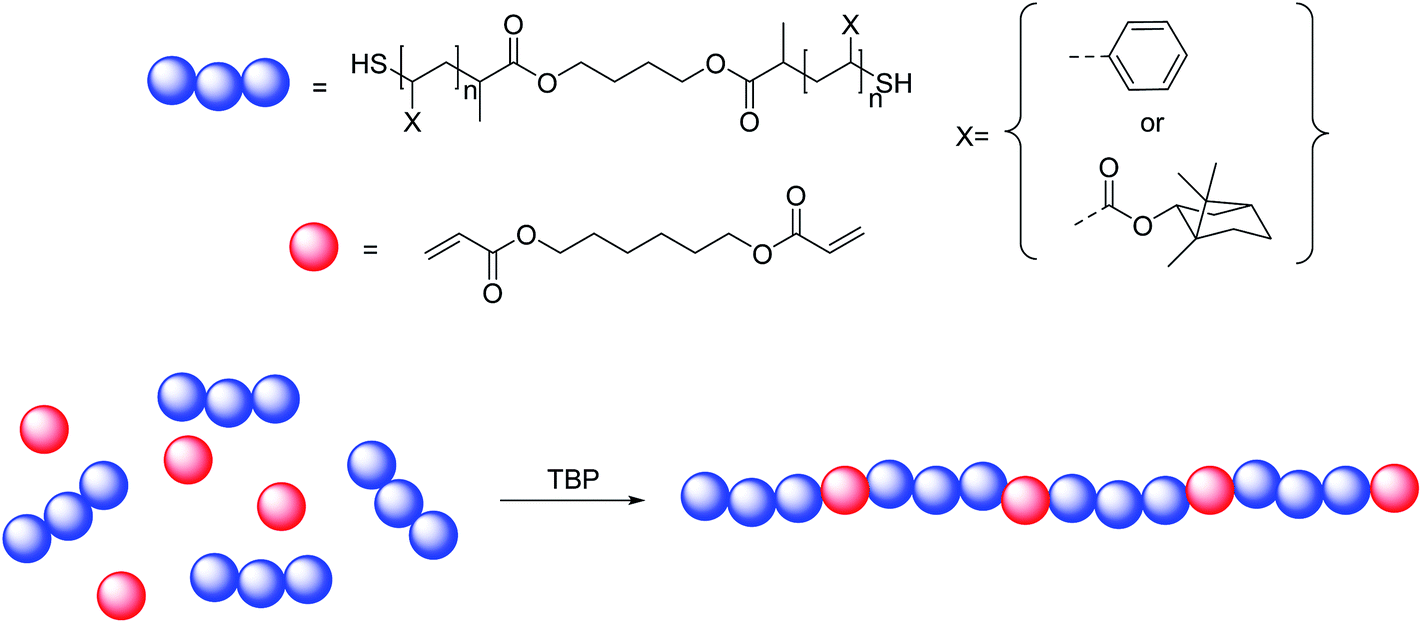 | ||
| Fig. 1 Schematic representation of the thiol–ene step growth polymerization of homotelechelic dithiol polymers and the diacrylate HDDA. | ||
In summary, the in here outlined synthesis strategy allows for generation of multi-segmented polymers, a class of materials with high potential with respect to their use in the build-up of complex macromolecular architectures. At the same time – and this is the main focus of this study – polymers are obtained that retain the physical characteristics of the underlying RAFT vinyl polymers, but which also incorporate the properties typically associated with PBTs, and which contain ester bonds in the backbone that are degradable in the sense that the multi-segmented linear chains or networks can be transformed back into the relatively low-molecular weight precursors. The multi-segmented PBT polymers are a type of poly(vinyl-co-ester) materials. Such polymers are often also synthesized by means of radical ring opening polymerization (RROP) and they are mainly designed in order to engineer degradability in polymer classes that otherwise are non-degradable at all.33 Depending on their design, poly(vinyl-co-ester)s combine properties such as degradability, non- or low-(cyto)toxicity, low glass transitions, specific viscosity and mechanical properties, even though their degradation products always contain short oligomeric chains of the underpinning non-degradable vinyl polymer. Poly(vinyl-co-ester)s find uses in recyclable plastics for waste management, but also in more biocompatible or biomedical applications. Our multi-segmented polymers are as such a worthwhile additional alternative to poly(vinyl-co-esters) prepared by RROP.
Experimental section
Materials
2,2′-Azobis(2-methylpropionitrile) (AIBN, Sigma-Aldrich, 98%) was recrystallized twice from ethanol prior to use, the monomers isobornyl acrylate (iBoA, Sigma-Aldrich, technical grade) and styrene (St, Acros, 99.5%) were deinhibited over a column of basic alumina prior to use. 2-([(Dodecylsulfanyl)carbonothioyl]sulfanyl)propanoic acid (DoPAT 1) RAFT-agent was synthesized according to a literature prodecure.34 2-Bromopropionic acid (Acros 99%), butanediol (Sigma-Aldrich, ≥97%), carbon disulfide (Acros, 99.9%), N,N′-dicyclohexylcarbodiimide (DCC, Acros, 99%), 4-(dimethylamino)pyridine (DMAP, Acros, 99%), dodecanethiol (Acros, 98%), hexanediol diacrylate (HDDA, Sigma-Aldrich, 80%), hexylamine (Acros, 99%), pentaerythritol tetraacrylate (PETA, Sigma-Aldrich, 10–40% triester), sodium hydroxide (NaOH, VWR), tetrapropylammonium bromide (Acros, 98%), tributylphosphine (TBP, Acros, 95%) and tris[2-(acryloyloxy)ethyl]isocyanurate (TAEIC, Sigma-Aldrich) were used as received. All solvents used were obtained from commercial sources (Acros, VWR, Sigma-Aldrich, p.a. grade) and used without further purification.Characterization
1H-NMR spectra were recorded in CDCl3 on a Varian Inova 300 spectrometer at 300 MHz using a 5 mm probe. FTIR spectra were collected with a Bruker Tensor 27 FT-IR spectrophotometer (nominal resolution 4 cm−1). UV-vis spectra were collected with an Agilent Cary 5000 UV-VIS-NIR spectrophotometer. Analytical SEC (Size Exclusion Chromatography) was performed on a Tosoh EcoSEC HLC-8320GPC, comprising an autosampler, a PSS guard column SDV (50 × 7.5 mm2), followed by three PSS SDV analytical linear XL (5 μm, 300 × 7.5 mm2) columns thermostated at 40 °C (column molecular weight range: 1 × 102 to 1 × 106 g mol−1), and a differential refractive index detector (Tosoh EcoSEC RI) using THF as the eluent at a flow rate of 1 mL min−1. Toluene was used as a flow marker. Analytical SEC (DMF GPC) was performed using a Spectra Series P100 (Spectra Physics) pump equipped with two mixed-B columns (10 μm, 2 cm × 30 cm, Polymer Labs) and a refractive index detector (Shodex) at 70 °C. DMF was used as the eluent at a flow rate of 1.0 mL min−1. Molecular weight distributions were determined relative to polystyrene standards because Mark–Houwink parameters are not available for the polymers under investigation. Electrospray ionization-mass spectroscopy (ESI-MS) was performed on an LTQ Orbitrap Velos Pro mass spectrometer (ThermoFischer Scientific) equipped with an atmospheric pressure ionization source operating in the nebulizer-assisted electrospray mode. The instrument was calibrated in the m/z range 220–2000 using a standard solution containing caffeine; MRFA, and Ultramark 1621. A constant spray voltage of 5 kV was used, and nitrogen at a dimensionless auxiliary gas flow rate of 5 and a dimensionless sheath gas flow rate of 10 were applied. The S-lens RF level, the gate lens voltage, the front lens voltage and the capillary temperature were set to 50%, −90 V, −8.5 V, and 275 °C respectively. A 250 μL aliquot of polymer solution with a concentration of 10 μg mL−1 was injected. A mixture of THF and methanol (THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 3:2), all HPLC grade, were used as solvent. Acrylic RAFT polymers were purified to remove monomer leftovers using a column filled with S-X1 beads from biobeads® (BIO-RAD laboratories) with a particle size between 200 and 400 mesh. DCM is the used eluent. Purification of acrylic dithiol polymers from dodecanethiol side products was performed on a recycling preparative HPLC LC-9210 NEXT system in the manual injection mode (3 mL) comprising a JAIGEL-1H and JAIGEL-2H column and a NEXT series UV detector using CHCl3 as the eluent with a flow rate of 3.5 mL min−1. Fractions were collected manually. TGA experiments were performed in a TA instruments TGA Q5000IR under nitrogen atmosphere (50 mL min−1). Sample masses were about 1–2 mg. The samples were first equilibrated at 60 °C, then heated to 700 °C (20 °C min−1) and subsequently cooled to 60 °C (50 °C min−1). DSC experiments were performed in a TA instruments DSC Q2000 under nitrogen atmosphere (50 mL min−1) using a LNCS cooling system. Sample masses (dried) were about 3–6 mg. The samples were dried overnight in open DSC crucibles (TA instruments aluminum Tzero DSC pans) in a vacuum oven at 60 °C under reduced pressure (0.6–0.8 bar) and subsequently sealed with a hermetic lid. In the DSC, the samples were first heated to 150 °C (20 °C min−1, 1 min iso), then cooled to −110 °C (10 °C min−1, 5 min iso) and subsequently heated a second time to 150 °C (20 °C min−1, 1 min iso). The second run was used to determine glass transition temperatures.
:MeOH = 3:2), all HPLC grade, were used as solvent. Acrylic RAFT polymers were purified to remove monomer leftovers using a column filled with S-X1 beads from biobeads® (BIO-RAD laboratories) with a particle size between 200 and 400 mesh. DCM is the used eluent. Purification of acrylic dithiol polymers from dodecanethiol side products was performed on a recycling preparative HPLC LC-9210 NEXT system in the manual injection mode (3 mL) comprising a JAIGEL-1H and JAIGEL-2H column and a NEXT series UV detector using CHCl3 as the eluent with a flow rate of 3.5 mL min−1. Fractions were collected manually. TGA experiments were performed in a TA instruments TGA Q5000IR under nitrogen atmosphere (50 mL min−1). Sample masses were about 1–2 mg. The samples were first equilibrated at 60 °C, then heated to 700 °C (20 °C min−1) and subsequently cooled to 60 °C (50 °C min−1). DSC experiments were performed in a TA instruments DSC Q2000 under nitrogen atmosphere (50 mL min−1) using a LNCS cooling system. Sample masses (dried) were about 3–6 mg. The samples were dried overnight in open DSC crucibles (TA instruments aluminum Tzero DSC pans) in a vacuum oven at 60 °C under reduced pressure (0.6–0.8 bar) and subsequently sealed with a hermetic lid. In the DSC, the samples were first heated to 150 °C (20 °C min−1, 1 min iso), then cooled to −110 °C (10 °C min−1, 5 min iso) and subsequently heated a second time to 150 °C (20 °C min−1, 1 min iso). The second run was used to determine glass transition temperatures.
Synthesis of the bifunctional RAFT agent BiDoPAT 2 (ref. 35)
DoPAT 1 (2 eq.) and 1,4-butanediol (1 eq.) were dissolved in DCM (15 mL/1 g DoPAT) in a three neck flask. The solution was stirred in an ice bath and continuously purged with nitrogen. DCC (2 eq.) and DMAP (0.1 eq.) were dissolved in DCM (about 1/3 of the volume used for the DoPAT/BD solution) and this mixture was subsequently added dropwise to the DoPAT/BD solution. After addition the ice bath was removed. The reaction mixture was stirred overnight at room temperature after which 100 mL H2O was added. The water phase was extracted three times with DCM. The recombined organic phases were dried over magnesium sulfate, filtered and the solvent was evaporated. The yellow solid product was purified by column chromatography [SiO2, n-hexane:ethyl acetate (1:1)] to yield the product 2 as a yellow solid (76%). 1H NMR (CDCl3): δ = 4.79 (q, 2H), 4.21–4.06 (m, 4H), 3.33 (t, J = 7.20 Hz, 4H), 1.75–1.60 (m, 8H), 1.58 (d, J = 7.4 Hz, 6H), 1.45–1.18 (m, 36H), 0.93–0.78 (m, 6H).
Polymerization of BiDoPAT-polystyrene 3a and 3b
AIBN (0.1 eq.), BiDoPAT (1 eq.) and styrene (40 eq.) were added in a vial. An equal volume of butyl acetate was added as solvent (50 vol%). The solution was sealed and flushed with N2 for 5 minutes. Afterwards the reaction vial was transferred into a glove box under inert atmosphere and placed in a pre-heated copper block of 80 °C for 6 hours. Afterwards, the reaction was quenched by removing the mixture from the glovebox and placing it in an ice bath. An NMR sample was taken to determine conversion. The polymer was precipitated in cold methanol and isolated. The final product 3a was analyzed using GPC. Mn = 2300 g mol−1, Đ = 1.17, conversionNMR = 41% (Fig. S2†). Also a second batch of BiDoPAT-PS 3b was prepared under the same conditions. Mn = 3000 g mol−1, Đ = 1.10.Polymerization of BiDoPAT-poly(isobornyl acrylate) (PiBoA) 4
Polymerization of iBoA was performed with AIBN (0.01 eq.), BiDoPAT (1 eq.) and iBoA monomer (10 eq.) in butyl acetate (50 vol%). The solution was sealed and flushed with N2 for 5 minutes. Afterwards the reaction vial was transferred into a glove box under inert atmosphere and placed in a pre-heated copper block of 80 °C for 90 min. Next, the reaction mixture was quenched in an ice bath. An NMR sample was taken to determine conversion. The reaction mixture was poured into an alumina pan to evaporate solvent and most monomer leftovers. Subsequently, the polymer was separated over a DCM wetted biobeads-SX1® column to purify the polymer from any residual monomer leftovers trapped inside the polymer matrix. The final purified polymer was analyzed using GPC and ESI-MS. Mn = 2300 g mol−1, Đ = 1.16, end group fidelity ≥ 95%, conversionNMR = 77% (Fig. S3†).Aminolysis into dithiol-PS 5
1 eq. of BiDoPAT-PS 3b was dissolved in DMF (75 vol%) and stirred at room temperature in ambient atmosphere. The aminolysis was started upon addition of 30 eq. of hexylamine. Furthermore, 1 eq. of TBP was added as reducing agent to avoid disulfide formation. After a reaction time of 30 min, the dithiol-PS 5 polymer was precipitated in cold methanol and isolated. Samples were analyzed both in THF-GPC and DMF-GPC. Furthermore, complete aminolysis was confirmed by 1H-NMR and FTIR. Mn = 2700 g mol−1, Đ = 1.18 (THF-SEC).Aminolysis into dithiol-PiBoA 6
1 eq. of BiDoPAT-PiBoA 4 was dissolved in acetone (75 vol%) and stirred at room temperature in ambient atmosphere. The aminolysis was started upon addition of 10 eq. of hexylamine. Furthermore, 1 eq. of TBP was added as reducing agent to avoid disulfide formation. After a reaction time of 2 hours, the reaction mixture was precipitated in cold methanol to isolate the crude polymer. Subsequently, the polymer was additionally purified by recycling GPC to obtain pure dithiol-PiBoA 6. Samples were analyzed with THF-GPC and ESI-MS. Mn = 1300 g mol−1, Đ = 1.78.Aminolysis and in situ thiol–ene (one-pot) polymerization towards HDDA-PS 7a1 and 7a2
For the one-step reaction, 1 eq. of BiDoPAT-PS 3a and 2.5 eq. of HDDA (corrected for its 80% purity) were dissolved in THF (75 vol%). The mixture was purged with nitrogen for 5 min and inserted into the glove box. Under this inert atmosphere, 5 eq. of hexylamine was added to start the reaction. In a second experiment, the same reagent mixture was prepared and the reaction was started upon addition of a combination of 5 eq. hexylamine and 5 eq. of TBP. Both reaction mixtures were stirred overnight after which the resulting polymers were precipitated in cold methanol. 7a1: Mn = 2600 g mol−1, Đ = 1.52, 7a2: Mn = 4100 g mol−1, Đ = 2.10.Thiol–ene step growth polymerization towards HDDA-PS 7b
1 eq. of dithiol-PS 5 and an equimolar amount (1.25 eq.) of HDDA (corrected for its 80% purity) were dissolved in THF (75 vol%). The reaction was started upon addition of 0.2 eq. of TBP. The mixtures were stirred at room temperature using varying reaction times. Afterwards, the reactions were quenched by precipitation in ice-cold methanol and the resulting polymers were isolated. Samples were analyzed by THF-GPC. Mn = 10700 g mol−1, Đ = 1.39.
Thiol–ene step growth polymerization towards HDDA-PiBoA 8
1 eq. of dithiol-PiBoA 6 and an equimolar amount (1.25 eq.) of HDDA (corrected for its 80% purity) were dissolved in acetone (75 vol%). The reaction was started upon addition of 0.2 eq. of TBP. The mixture was stirred at room temperature for 30 min. Afterwards, the reaction was quenched by precipitation in ice-cold methanol and the resulting HDDA-PiBoA 8 polymer was isolated. The polymer was analyzed by THF-GPC. Mn = 3300 g mol−1, Đ = 1.81.Thiol–ene step growth polymerization towards branched polymers and networks 9–14
The synthesis of branched polymers 9–14 was performed using either tris[2-(acryloyloxy)ethyl]isocyanurate (TAEIC) or pentaerythritol tetraacrylate (PETA) as acrylate cross-linker. These compounds were mixed with the linear HDDA in different ratios. Dithiol-PS 5 was added in such amounts that equimolar thiol and acrylate functionalities were present. Reactions were performed at room temperature in ambient atmosphere. 75 vol% of THF (compared to dithiol-PS) was used as solvent and 0.2 eq. of TBP was added to initiate the polymerization. The reaction mixtures were stirred for 2–3 hours. The resulting branched polymers and networks were precipitated in cold methanol and isolated by decantation of the solvent.Enzymatic degradation of HDDA-PS 7b
2 mg of HDDA-PS 7b and 0.2 mg porcine liver esterases (PLE, activity 15 U per mg solid, with U representing the hydrolysis of 1.0 μmol of ester to its respective acid and alcohol per minute at pH = 8.0 and T = 37 °C) were added to 1.5 mL phosphate buffered saline (PBS, pH 7.4) in an Eppendorf tube. The Eppendorf was placed on a temperature controlled shaker of 37 °C and shaken for 8 days. The degraded polymer was extracted with chloroform and dried in vacuo. The degradation products were analyzed with THF-SEC. Mn = 1100 g mol−1, Đ = 5.94 (multimodal distribution).Basic degradation of HDDA-PS 7b
10 mg of HDDA-PS 7b was dissolved in 1.5 mL THF. Then 1.5 mL of an aqueous basic solution of NaOH (1 M) was added. The mixture was stirred for 4 days at 40 °C to induce basic hydrolysis of the ester bonds. The solution was then neutralized with HCl (1 M) and the degraded polymer was subsequently extracted with DCM. The degradation products were analyzed with THF-SEC. Mn = 1600 g mol−1, Đ = 1.37 (bimodal distribution) and Mn = 200 g mol−1, Đ = 1.05.Results and discussion
RAFT polymerizations
As a first step in the synthesis of bifunctional telechelic precursor polymers, a bifunctional RAFT agent BiDoPAT 2 was prepared (Scheme 1) by reacting DoPAT with butanediol via Steglich esterification. Subsequently, this chain transfer agent was employed in the RAFT polymerizations of styrene and isobornyl acrylate (iBoA). Two batches of BiDoPAT-PS were prepared, 3a (Mn = 2300 g mol−1, Đ = 1.17) and 3b (Mn = 3000 g mol−1, Đ = 1.10) and one batch of BiDoPAT-PiBoA 4 (Mn = 2300 g mol−1, Đ = 1.16). Full monomer conversions were not targeted in order to avoid the progressive loss of active trithiocarbonate end groups of the polymer, which would lower further step-growth efficiency. The acrylic polymer was further characterized by means of soft ionization mass spectrometry (ESI-MS, see Fig. 2 top) to reveal high end group fidelity (≥95%). Both single charged and double charged BiDoPAT-PiBoA species were observed in the mass spectrum (see Scheme 2 and Table S1†). | ||
| Scheme 1 Synthetic pathway towards thiol–ene step-growth polymers and networks from aminolysed bifunctional RAFT precursor polymers. | ||
 | ||
| Fig. 2 ESI-MS spectra of BiDoPAT-PiBoA 4 (top) and dithiol-PiBoA 6 (bottom). Left: full MS spectra. Right: zoom into a single monomer repeating unit. | ||
 | ||
| Scheme 2 Structures of main product species observed in ESI-MS spectra. | ||
Aminolysis and (in situ) thiol–ene step growth into multi-segmented polymers
After synthesis of the bifunctional RAFT polymer precursors, the trithiocarbonate RAFT end groups had to be converted into thiols to facilitate a Michael addition with a diacrylate into multi-segmented thiol–ene step growth polymers. Since both reactions can be executed by means of a primary amine, two reaction pathways can in principle be considered; the first synthesis route comprises the aminolysis and in situ thiol–ene step growth reaction in a one-pot procedure. This route has the advantage that no intermediate thiol polymers need to be isolated. Furthermore, the undesired formation of disulfide bridges is reduced since any formed thiol functionality can react instantaneously with the diacrylate present. To this end, 1 equiv. BiDoPAT-PS 3a and hexane dioldiacrylate (HDDA) were dissolved in THF. In order to optimize the success of the aminolysis and subsequent thiol–ene reaction, a few important issues must, however, be addressed. First of all, two equivalents of hexylamine are needed to fully reduce one trithiocarbonate moiety, so four equivalents are required in order to convert the bifunctional BiDoPAT polymer precursor into homotelechelic dithiol polymer (see Scheme 3). Furthermore, when the trithiocarbonate RAFT end groups are aminolyzed with hexylamine, not only the desired dithiol polymer is formed, but also dodecanethiol evolves as a side product, which can also react with HDDA in the in situ thiol–ene reaction, thereby disrupting the stoichiometric thiol to acrylate ratio necessary for optimal step-growth polymerization. Furthermore, the dodecanethiol side chains will inevitably end cap the growing step-growth polymer and hence limit the achievable molecular weight. In order to account for this effect, 2.5 equivalents of HDDA (to account for the 80% purity of HDDA) were added with respect to 1 equiv. of obtained dithiol-PS 5. To ensure complete aminolysis and in situ thiol–ene Michael addition, 5 equiv. of hexylamine were employed. The reaction was carried out under inert atmosphere (to avoid disulfide reactions) and stirred at room temperature overnight, after which the resulting polymer HDDA-PS 7a1 was isolated by precipitation in cold MeOH. The polymer was analyzed by THF-SEC (Fig. 3, left). No substantial shift of the chromatogram was observed, indicating failure of reaction. In a second attempt, the reaction was carried out by using a combination of 5 equiv. hexylamine and 5 equiv. of tributyl phosphine (TBP). This is a strong nucleophile that is able to catalyze the thiol–ene reaction very efficiently and furthermore it is a reducing agent that breaks up any formed disulfide bridges. Again the reaction was stirred overnight and the resulting HDDA-PS 7a2 was precipitated in MeOH. In this case, a clear shift of the chromatogram towards higher molar masses was observed, indicating successful formation of a multi-segmented polymer. The dispersity of polymer increased to 2.10, a value characteristic for step-growth polymers (Table 1). The increase in number average molar mass suggests that about 2 to 3 dithiol-PS chains were coupled in the final HDDA-PS 7a2. | ||
| Scheme 3 Overview of formation of dodecanethiol and zwitter-ionic side-products that inevitably lead to end-capping of the HDDA-PS step-growth polymer during a one-pot procedure. | ||
 | ||
| Fig. 3 Molar mass distributions of BiDoPAT-PS 3a and 3b RAFT precursors and HDDA-PS thiol–ene step growth polymers. Left: synthesized via one-pot aminolysis and in situ thiol–ene polymerization (7a1 and 7a2). Right: synthesized via 2-steps, first aminolysis into dithiol-PS 5, followed by thiol–ene polymerization (7b), analyzing increasing reaction times. | ||
| Polymer | M n (g mol−1) | Đ | M w (g mol−1) |
|---|---|---|---|
| BiDoPAT-PS 3a | 2300 | 1.17 | 2700 |
| HDDA-PS 7a1 | 2600 | 1.52 | 3900 |
| HDDA-PS 7a2 | 4100 | 2.10 | 8600 |
| BiDoPAT-PS 3b | 3000 | 1.10 | 3300 |
| Dithiol-PS 5 | 2700 | 1.18 | 3100 |
| HDDA-PS 7b | 10700 |
1.39 | 14800 |
| BiDoPAT-PiBoA 4 | 2300 | 1.16 | 2600 |
| Dithiol-PiBoA 6 | 1300 | 1.78 | 2400 |
| HDDA-PiBoA 8 | 3300 | 1.81 | 6100 |
In the second synthetic route towards multi-segmented thiol–ene polymers from bifunctional RAFT precursors, aminolysis and thiol–ene step-growth reaction were performed in two separate steps, with intermediate purification of the dithiol polymer in between. This way, the undesired dodecanethiol side products can be removed before the purified dithiol polymer reacted further with HDDA. Furthermore, it allows to drastically reduce the amount of nucleophilic/base catalyst required for the thiol–ene reaction. It is known from literature that a nucleophilic catalyst such as hexylamine or TBP reacts with the diacrylate via a zwitter-ionic structure.21 When formed in catalytic amounts, this zwitter-ion quickly reacts with a thiol yielding a thiolate anion, which in its turn reacts with another diacrylate molecule, releasing the catalyst to complete the catalytic cycle. However, when larger amounts of catalyst are used, too high concentrations of the zwitter-ionic species are formed which, as undesired amine side products, decrease the efficiency of the thiol–ene step-growth reaction (Scheme 3). Therefore, in all our further experiments, we first performed the aminolysis reaction, purified the obtained dithiol polymer, and subsequently executed the thiol–ene reaction.
To obtain dithiol-PS 5, 1 equiv. of BiDoPAT-PS 3b was dissolved in DMF and reacted with 30 equiv. of hexylamine and 1 equiv. of TBP to insure fast and efficient aminolysis of the trithiocarbonate end groups. The resulting polymer was precipitated in cold MeOH and analyzed via THF-SEC. A small shift towards lower molar mass is observed (Fig. 3, right), corresponding with the loss of the trithiocarbonate end groups (−488 Da for each end group). Furthermore, a shoulder on the high molar mass side of the chromatogram indicated formation of some disulfide bridges. Since reactions were carried out in the presence of a reducing agent (TBP), the sulfur bridges are believed to be formed during SEC separation – most likely caused by the presence of peroxides in the THF. This hypothesis was supported by analysis of the molar mass distribution of dithiol-PS 5 using DMF-SEC (see Fig. S1†). Although the distribution appears somewhat broader than in THF-SEC (due to differences in hydrodynamic volume), no high molar mass shoulder is present. Next, the end group conversion was investigated. Since PS is difficult to analyze in ESI-MS, the dithiol-PS 5 was characterized by 1H-NMR and FTIR and compared to the initial BiDoPAT-PS 3b before aminolysis (Fig. 4 left and right). The NMR spectrum shows the disappearance of the resonances around 1.3 ppm (BiDoPAT alkyl chain) and 3.1–3.3 ppm (–S(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)S–C
S)S–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–), demonstrating complete removal of the RAFT end groups. This is also supported by the FTIR spectra, which show disappearance of the vibration bands at 810 cm−1 and 1067 cm−1 (both characteristic for the trithiocarbonate end groups) and appearance of a broad band around 2650–2500 cm−1, characteristic for thiols. Finally, the UV-vis spectrum of non-colored dithiol-PS 5 showed disappearance of the trithiocarbonate absorption between 380 and 500 nm, when compared to the spectrum of the yellow colored BiDoPAT-PS 3b (see Fig. S4†).
2–), demonstrating complete removal of the RAFT end groups. This is also supported by the FTIR spectra, which show disappearance of the vibration bands at 810 cm−1 and 1067 cm−1 (both characteristic for the trithiocarbonate end groups) and appearance of a broad band around 2650–2500 cm−1, characteristic for thiols. Finally, the UV-vis spectrum of non-colored dithiol-PS 5 showed disappearance of the trithiocarbonate absorption between 380 and 500 nm, when compared to the spectrum of the yellow colored BiDoPAT-PS 3b (see Fig. S4†).
 | ||
| Fig. 4 1H-NMR (left) and FTIR (right) spectra of BiDoPAT-PS 3b and dithiol-PS 5, after aminolysis. The NMR resonances marked with an M correspond to monomer leftovers trapped inside the BiDoPAT-PS 3b polymer matrix and the resonances marked with an asterisk result from leftover of CHCl3, MeOH and DMF solvents in the dithiol-PS 5 matrix. | ||
Dithiol-PS 5 was then reacted with an equimolar amount of HDDA (1.25 equiv. to account for 80% purity of HDDA) in the subsequent thiol–ene reaction, using 0.2 equiv. of TBP at room temperature. Samples were quenched in MeOH after different reaction times and analyzed via THF-SEC (Fig. 3, right). Upon increasing reaction times until 60 min, a clear shift of the distribution towards higher molar masses is observed. The number average molar mass of the final carefully precipitated multi-segmented HDDA-PS 7b is 10700 g mol−1, with a dispersity of 1.39 (Table 1). About 4 to 5 dithiol-PS chains are connected on average in the final step-growth polymer. This number of connected segments is very similar to values of a broad variety of multi-segmented polymers reported recently in literature.36–38 The value may appear low, but considerable tailing towards high molecular weights is observed, with peak molecular weights well above 104 Da. Thus, multi-segmented polymers with considerable molecular weight are obtained in the process and materials with higher average molar masses are in principle obtainable by additional selective precipitation purification. Furthermore, a further increase in number of coupled segments and final molar mass might be expected when HDDA with a higher purity grade could be used in future. The commercially available HDDA has a purity of around 80%. Although we attempted to correct the stoichiometric ratio for this impurity (by using 1.25 equiv. of HDDA), still the thiol:acrylate ratio might be not perfectly equimolar, lowering the success of step-growth coupling to some extent. Nevertheless, by comparing the two-step with the one-pot reaction, a clear improvement of the reaction outcome is seen, demonstrating the better efficiency of the two-step reaction.
Next to the synthesis of linear HDDA-PS 7b, also cross-linked thiol–ene polymers were targeted using either tris[2-(acryloyloxy)ethyl]isocyanurate (TAEIC) or pentaerythritol tetraacrylate (PETA) as acrylate cross-linker (Scheme 1). Different mixtures of TAEIC/HDDA and PETA/HDDA were prepared in THF and dithiol-PS 5 was added in such amounts that equimolar thiol and acrylate functionalities were achieved. Upon addition of 0.2 equiv. TBP, the thiol–ene polymerization is conveniently started. After stirring for 2–3 hours at room temperature the precipitated networks were isolated in cold MeOH. The materials prepared with only multifunctional acrylates, PETA-PS 11 and TAEIC-PS 14, appear as white brittle insoluble networks. The networks containing 50% or 20% cross-linker also did not fully dissolve: only low molar mass fractions of the material were able to be brought in solution. All polymer networks are, however, studied by means of TGA and DSC to characterize their thermal behavior (see below).
Next to the PS precursors, it was investigated whether also other polymer precursors can be coupled via thiol–ene reactions. To this end, BiDoPAT-PiBoA 4 was used as precursor polymer for the aminolysis and subsequent thiol–ene step-growth polymerization with HDDA. Initial attempts to aminolyze the end groups of the bifunctional PiBoA RAFT precursor failed. ESI-MS revealed that some of the obtained thiol end groups had reacted with leftovers of iBoA monomer that were still present in the polymer matrix and hence had irreversibly endcapped the precursor. In the above described aminolysis of PS precursors, leftovers of styrene monomers are unable to react with the dithiol polymer since the vinyl bond of styrene is not electron deficient and therefore is unable to undergo Michael addition. To overcome this problem, BiDoPAT-PiBoA 4 had to be purified thoroughly in order to remove all residual monomer. As precipitation procedures are only able to remove about 95% of remaining iBoA, the polymer was purified further via column chromatography on a BioBeads S-X1 column, which is able to separate molecules based on their hydrodynamic volume. Like in a SEC column, the larger polymer molecules elute first, while the small monomer molecules are retained much longer. In this way, purified BiDoPAT-PiBoA 4 polymer was obtained. The RAFT precursor was then dissolved in acetone and reacted with 10 equiv. hexylamine and 1 equiv. TBP to obtain crude dithiol-PiBoA 6. Again undesired dodecanethiol side chains were formed as side products and the polymer had to be further purified to remove excess hexylamine before executing the subsequent thiol–ene reaction with HDDA. The crude polymer was precipitated in cold MeOH. Unfortunately, this purification procedure proved to be insufficient since further attempts to perform the thiol–ene reaction on this material failed to yield significantly increased molar mass material. Therefore, the precipitated dithiol-PiBoA 6 was purified additionally to remove dodecanethiol and base leftovers by means of recycling SEC, another technique commonly used to separate molecules with different molar masses from each other.39 The purified dithiol-PiBoA 6 was then analyzed by ESI-MS to reveal that aminolysis was mostly successful (Fig. 2, bottom). Both single and double sodium charged dithiol-PiBoA species were observed as main distribution in the mass spectrum (Scheme 2). Also some additional minor peaks were observed not corresponding to dithiol-PiBoA and of which the origin at the moment is unclear. Furthermore, it must be noted that in some measurements a peak series with a m/z value of −2 Da was observed, as compared to the dithiol-PiBoA species. This corresponds to a disulfide product whereby two dithiol-PiBoA species have coupled. Since for the ESI-MS analysis samples are dissolved in THF and heated to 275 °C during injection in the mass spectrometer, it is possible that also here the disulfide bridges are formed in situ during measurement. Furthermore, a high molar mass shoulder resulting from in situ formation of disulfides is once more observed in the THF-SEC chromatograms of dithiol-PiBoA 6 (Fig. 5) as was also described in the previous section for the dithiol-PS product. The purified dithiol-PiBoA 6 polymer was reacted with HDDA in the presence of 0.2 equiv. TBP to obtain step-growth polymer HDDA-PiBoA 8 that was isolated by precipitation in cold MeOH. An obvious shift of the molar mass distribution is observed, which is, however, less pronounced than what was obtained for HDDA-PS 7b (Fig. 5). The dispersity of polymer increased to 1.81 and the increase in number average molar mass suggests that about 2 to 3 dithiol-PiBoA chains are on average conjugated in the final HDDA-PiBoA 8. In addition to the stoichiometric imbalance already created by the impurities in the applied HDDA, additional errors in the dithiol:acrylate ratio are created by the minor part of the unidentified PiBoA polymer distribution which does not contain dithiol end groups. This fact most likely explains the lower step-growth efficiency for the HDDA-PiBoA thiol–ene polymer, compared to the HDDA-PS material. To conclude, it is demonstrated that the synthesis of thiol–ene polymers based on bifunctional RAFT precursors is also possible for PiBoA, however, it is of crucial importance to purify both RAFT as well as dithiol PiBoA polymers thoroughly in order to have a successful thiol–ene conjugation reaction in the end.
 | ||
| Fig. 5 Molar mass distributions of BiDoPAT-PiBoA 4 RAFT precursor, dithiol-PiBoA 6 aminolysis product and HDDA-PiBoA 8 thiol–ene step growth polymer. | ||
Thermal characterization
The thermal stability of the linear multi-segmented HDDA-PS 7b was investigated by means of thermogravimetric analysis (TGA, Fig. 6). It can be seen from Fig. 6 that HDDA-PS exhibits a minor weight loss of 2% around 125–230 °C, which is attributed to the loss of unreacted HDDA monomer. The major degradation step only occurs around 350–450 °C, demonstrating the good thermal stability of HDDA-PS 7b. Subsequently, the polymer was characterized by differential scanning calorimetry (DSC) in order to determine the glass transition temperature Tg (Fig. 7, left). The Tg seems to follow a two-step process: the heat flow curves show a gradual onset from −50 °C up to (close to) the midpoint (nearly 50% of the glass transition step), followed by a second sharp step (less than 20 °C wide). It is well-known from literature that linear PBT polymers obtained from HDDA and small thiols such as butane dithiol, exhibit Tg values between −80 °C and −60 °C.40,41 On the other hand, the Tg of normal PS lies around 100 °C. Since for the current HDDA-PS 7b dithiols from PS precursor polymers are used in combination with small size HDDA, a glass transition around an intermediate temperature of 45 °C is not unexpected (Table 2). It can thus be concluded that the Tg of a PBT polymer can be tuned based on the type and ratio of polymeric vs. small molecule thiol and acrylate units. This certainly opens new possibilities towards the design, development and implementation of such PBT materials in a variety of future applications. | ||
| Fig. 6 TGA weight loss (left) and derivative weight loss (right) profiles for linear HDDA-PS 7b thiol–ene polymer and thiol–ene networks 9–14. | ||
 | ||
| Fig. 7 DSC results showing second heating curve for HDDA-PS 7b linear thiol–ene polymer (left) and networks 9–14 (right). | ||
| Polymer | T g (°C) |
|---|---|
| HDDA-PS 7b | 45 |
| HDDA-PETA0.2-PS 9 | 43 |
| HDDA-PETA0.5-PS 10 | 52 |
| PETA-PS 11 | 51 |
| HDDA-TAEIC0.2-PS 12 | 28 |
| HDDA-TAEIC0.5-PS 13 | 51 |
| TAEIC-PS 14 | 43 |
| PS | ∼100 |
| BDDA-PETMP | −31 |
| BDDA-PETMP0.1-BDT | −58 |
| BDDA-TMPEIC0.1-BDT | −100 |
Furthermore, also the thermal stability of the cross-linked PS thiol–ene networks 9–14 was investigated by means of TGA (Fig. 6). Again most networks exhibit minor weight losses (2–8%) between 125–260 °C, which are attributed to the loss of unreacted residual monomer or cross-linker. Furthermore, network 9 exhibits a 13% weight loss at a temperature range as low as 70–120 °C. It is assumed that this network may have retained some THF solvent or absorbed some water before analysis. The main degradation of the networks occurs between 300–500 °C, the same temperature range at which PBT networks consisting of small molecular weight dithiols and diacrylates disintegrate.29 As expected, also the current thiol–ene networks made from polymeric dithiols and small acrylates display a good thermal stability. The networks were also characterized by DSC (Fig. 7, right). In our previous report on PBT networks from solely small molecules, in all cases sub-zero glass transition temperatures were reported (−60 °C to −28 °C). In the current case, the PBT networks are built from a combination of polymeric dithiol-PS precursors and small size molecules such as HDDA monomers and PETA or TAEIC cross-linkers. The current multi-segmented PS networks thus exhibit Tg values around intermediate temperature ranges (Table 2), around room temperature to 50 °C, as was also observed above for the linear HDDA-PS polymer. Furthermore, the DSC profiles show that also for the networks a two-step process is observed for Tg. This indicates that the networks may have a (partially)phase-segregated structure (phases at least larger than 1 nm) with domains having a quite uniform and higher cross-link density, giving the sharp step, and domains with a lower, varying cross-link density (possibly a gradient), yielding the gradual onset. Furthermore, there is no increasing trend of Tg upon increasing the cross-linker concentration. This indicates the thiol–ene cross-linking reaction is most likely not completed due to steric hindrance becoming too strong for the last functional groups of the multifunctional cross-linkers. Another explanation is that internal cyclization of some of the cross-linking units occurs. One other complicating factor in the analysis is the purification of the materials, which is easier for the linear polymer than for the networks. A few wt% of small molecules (e.g., HDDA) remaining in the network structure will have a considerable plasticizing effect, lowering the observed Tg. On the other hand, recent studies on thiol–ene polymers have reported that upon ageing, oxidation of the sulfide linkages into sulfoxides and sulfones, alters the material properties.42 Oxidation of the networks over time leads to more brittle and stiff materials, with a higher glass transition temperature. All the above described factors have an influence on the observed glass transitions and have to be taken into consideration.
Polymer degradation
All multi-segmented thiol–ene polymers synthesized in this study contain ester bonds at each HDDA linkage and also in the middle of the employed dithiol polymer backbone (see Scheme 1). The ester bonds are sensitive to hydrolysis and the thiol–ene materials are therefore susceptible to degradation. As a representative example the (bio)degradability of HDDA-PS 7b was investigated. In a first test, the polymer was added to a solution of porcine liver esterase (PLE) in PBS buffer (pH 7.4). The polymer was shaken in this mixture for 8 days at 37 °C. Afterwards, the degraded material was recuperated by extraction with an organic solvent and analyzed with THF-SEC. The results of the enzymatic degradation are displayed in Fig. 8, green line. As can be seen from the figure, a period of 8 days is insufficient to fully degrade the HDDA-PS 7b into its individual PS segments. The molar mass distribution of the initial multi-segmented polymer is mostly still present. However, a broad peak at low molar mass (≤1000 g mol−1) arises, which indicates that degradation occurred to some extent. Since HDDA-PS 7b is insoluble in the applied PBS buffer, only the outer sphere of the polymer aggregates in the dispersion can get in contact with the enzyme and hence only surface degradation will occur. | ||
| Fig. 8 Molar mass distributions of HDDA-PS 7b, before and after degradation using porcine liver esterase (PLE in PBS, green line) or NaOH (1 M) H2O/THF solution (pink line). Also the molar mass distribution of dithiol-PS 5 is given as reference. | ||
To test if faster degradation can be reached by other means, HDDA-PS 7b was dissolved in a small amount of THF. Subsequently, an equal volume of a 1 M NaOH solution in water was added. The resulting mixture was stirred for 4 days at 40 °C and then neutralized by addition of HCl (1 M). The degraded material was extracted with DCM and analyzed with THF-SEC (Fig. 8, pink line). This time, a full degradation of the material is observed. In theory, when all available ester bonds in the polymer backbone are hydrolyzed, the remaining polymeric fragments should have a molar mass of half the size of the dithiol-PS 5, because the initial BiDoPAT agent also contains two hydrolysable ester bonds. In the SEC of the degraded material, 3 distinct distributions can be observed. The first one, at around 3000 Da corresponds to the initial dithiol-PS fragments. A second distribution at around 1500 Da matches with fragments where the original dithiol-PS was cut in half by the BiDoPat degradation. The third distribution around 300 Da is assumed to originate from degraded hexanediol fragments (from HDDA). This degradation experiment under alkaline conditions clearly demonstrates that the thiol–ene polymers presented in this study can be degraded over time, either within a timeframe of 4 days via strong basic conditions (where base can penetrate the networks), or via enzymatic surface degradation, which is accordingly much slower due to the mechanistic differences in the degradation process. This opens the pathway for these multi-segmented poly(vinyl-co-ester) architectures towards prospective ecological or biomedical applications for which (bio)degradability is required. It is though very important to stress that the underlying vinyl polymer segment is obviously non-degradable, only the linkages can be broken. Yet, small fragments of few hundred Da are easily removed from a system compared to the large molecular weight counterparts in the multi-segment network materials.
Conclusions
Multi-segmented polymer materials with interesting properties with respect to glass transition and degradability have been obtained from a combination of RAFT polymerization and a Michael addition β-thioester linking reaction. Thiol–ene Michael addition for the formation of multi-segmented and (bio)degradable polymers has not been studied in depth before, thus this study also fills a significant gap in the applicability of thiol–ene for the generation of complex macromolecular architectures. Bifunctional telechelic BiDoPAT-PS 3 and BiDoPAT-PiBoA 4 precursor polymers were successfully synthesized via RAFT using a bifunctional CTA agent. The polymers display narrow molar masses distributions and high end group fidelities (≥95%). The trithiocarbonate polymer end groups are then aminolyzed into thiol functionalities. The success of the end group modification is demonstrated by 1H-NMR and FTIR for the dithiol-PS 5 and by ESI-MS for the dithiol-PiBoA 6. In a one-pot reaction, the obtained dithiol polymers can be coupled in situ with HDDA into step-growth thiol–ene polymers. However, this reaction is only partially successful due to interfering side reactions that end cap the polymers and impede further step-growth. Best results are obtained when the dithiol polymers are first purified after aminolysis in order to remove dodecanethiol side products, monomer leftovers and excesses of hexylamine. The isolated dithiol-PS 5 and dithiol-PiBoA 6 are then polymerized in a step-growth fashion with HDDA via phosphine catalyzed thiol–ene Michael additions into HDDA-PS 7b and HDDA-PiBoA 8. On average, about 4–5 dithiol-PS chains and 2–3 dithiol-PiBoA chains are conjugated with HDDA linkages in the final multi-segmented polymers, demonstrating superior results for the PS based PBT materials. All distributions of the multi-segmented polymers are broadened towards high molar masses, indicating that also chains with significantly higher numbers of segments are formed alongside in the process.Besides linear polymers, dithiol-PS 5 was also reacted with multifunctional acrylates, obtaining high-molecular weight PBT polymer networks. Both linear HDDA-PS 7b as well as the PS based insoluble networks 9–14 were characterized by TGA and DSC. All materials display good thermal stabilities with major degradation occurring around 300–500 °C and glass transitions around 30–50 °C that lie in between the Tg of PS (100 °C) and the sub-zero Tg's typically obtained for PBT networks obtained from small thiol and acrylate monomers. The strategy to use RAFT polymers as PBT building blocks allows thus to create materials with tunable thermal properties that retain to some degree the characteristics of the underlying vinyl polymer. Finally, the (bio)degradability of the multi-segmented HDDA-PS 7b towards its individual PS segments was tested by alkaline or enzymatic (PLE) hydrolysis of the labile ester bonds in the PBT polymer backbone. Enzymatic degradation by PLE was tested for a period of 8 days. Surface degradation of the hydrophobic material is observed, leading to degradation on a larger time scale. Degradation of the polymer by means of NaOH in a water/THF mixture is considerably faster and the multi-segmented HDDA-PS 7b fully degrades into very small polymeric segments within just 4 days.
This type of degradable multi-segmented polymer materials could on one the hand be employed for the development of more green plastics, whereby a classical polymeric precursor backbone can be chosen based on desired properties. On the other hand, due to their easily tunable properties and their ability to degrade, they are also considered as potential candidates for more high-end (bio)medical applications. First tests on biocompatibility of PBTs are encouraging. Either way, this type of multi-segmented thiol–ene materials based on polymeric precursors constitutes a new and interesting class of biodegradable polymers with an interesting future for prospective appliances.
Acknowledgements
This study was a part of the Interreg IV-A project “BioMiMedics” (http://www.biomimedics.org). In the framework of Interreg IV-A, the financial contribution from the EU and the province Limburg (Belgium) is kindly acknowledged. The authors are also grateful for funding via a postdoctoral mandate of J. V. from the Fonds Wetenschappelijk Onderzoek (FWO). Additionally, support from Belgian Science Policy in the frame of IAP-PAI P7/05 and the Hercules foundation for funding in the framework of the project “LC-MS@UHasselt: Linear Trap Quadrupool-Orbitrap mass spectrometer” is gratefully acknowledged.References
- A. K. Mohanty, M. Misra and G. Hinrichsen, Macromol. Mater. Eng., 2000, 276/277, 1–24 CrossRef CAS.
- G. E. Luckachan and C. K. S. Pillai, J. Polym. Environ., 2011, 19, 637–676 CrossRef CAS.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- M. A. Meyers, P.-Y. Chen, A. Y.-M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53, 1–206 CrossRef CAS.
- I. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- S. Kobben, A. Ethirajan and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1633–1641 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- J. W. Chan, H. Wei, H. Zhou and C. E. Hoyle, Eur. Polym. J., 2009, 45, 2717–2725 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. J. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Muller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- C. Boyer, G. Boutevin, J.-J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 395–415 CrossRef CAS.
- C. Boyer, G. Boutevin, J.-J. Robin and B. Boutevin, Polymer, 2004, 45, 7863–7876 CrossRef CAS.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- C. Gimbert, M. Moreno-Manas, E. Perez and A. Vallribera, Tetrahedron, 2007, 63, 8305–8310 CrossRef CAS.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS.
- J. Vandenbergh, K. Ranieri and T. Junkers, Macromol. Chem. Phys., 2012, 213, 2611–2617 CrossRef CAS.
- N. Zaquen, B. Wenn, K. Ranieri, J. Vandenbergh and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 178–187 CrossRef CAS.
- J. Vandenbergh, M. Peeters, T. Kretschmer, P. Wagner and T. Junkers, Polymer, 2014, 55, 3525–3532 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- E. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2118–2126 CrossRef CAS.
- P. L. Golas, N. V. Tsarevsky, B. S. Sumerlin, L. M. Walker and K. Matyjaszewski, Aust. J. Chem., 2007, 60, 400–404 CrossRef CAS.
- F. Driessen, F. E. Du Prez and P. Espeel, ACS Macro Lett., 2015, 4, 616–619 CrossRef CAS.
- J. Vandenbergh, G. Reekmans, P. J. Adriaensens and T. Junkers, Chem. Commun., 2013, 49, 10358–10360 RSC.
- J. Shin, H. Matsushima, J. W. Chan and C. E. Hoyle, Macromolecules, 2009, 42, 3294–3301 CrossRef CAS.
- S. Tomassi, R. Bizzarri, R. Solaro and E. Chiellini, J. Bioact. Compat. Polym., 2002, 17, 3–21 CrossRef.
- F. Goethals, S. Martens, P. Espeel, O. van Den Berg and F. E. Du Prez, Macromolecules, 2014, 47, 61–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Molar mass chromatogram of dithiol-PS 5, measured by DMF-SEC, 1H-NMR spectra of BiDoPAT-PS 3a and BiDoPAT-PiBoA 4 (before purification from residual monomer to determine conversion), UV-vis spectra of BiDoPAT-PS 3a and dithiol-PS 5 and a table of m/z values for products identified in the ESI-MS spectra. See DOI: 10.1039/c5ra18861j |
| This journal is © The Royal Society of Chemistry 2015 |