
The facile synthesis, characterization and evaluation of photocatalytic activity of bimetallic FeBiO3 in natural sunlight exposure
M. Aslama,
M. Tahir Soomroa,
Iqbal M. I. Ismailab,
Huda A. Qaric,
M. A. Gondald and
A. Hameed*ae
aCentre of Excellence in Environmental Studies (CEES), King Abdulaziz University, Jeddah 21589, Kingdom of Saudi Arabia. E-mail: afmuhammad@kau.edu.sa; hameedch@yahoo.com
bChemistry Department, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Kingdom of Saudi Arabia
cDepartment of Biological Sciences, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
dDepartment of Physics, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
eNational Centre for Physics, Quaid-e-Azam University, Islamabad 44000, Pakistan
First published on 26th November 2015
Abstract
In an effort to develop sunlight active photocatalysts for environmental remediation, a phase pure bimetallic oxide, FeBiO3, was synthesized by a facile route. The optical, structural and morphological properties of the synthesized FeBiO3 were compared with both the parent oxides i.e. α-Fe2O3 and Bi2O3. The synthesized powder exhibited strong absorption in the visible region with the appearance of a distinct optical absorption edge at 2.15 eV. The PL, Raman and IR spectroscopic investigations of the synthesized powder, in comparison to that of pure oxides, confirmed the formation of pure phase FeBiO3 and revealed the dominant role of Fe3+ ions in controlling the optical properties of the material. The X-ray diffraction analysis (XRD) revealed the hexagonal or rhombohedral geometry with an average crystallite size of 20.2 nm, whereas the X-ray photoelectron spectroscopy (XPS) verified the existence of both Fe and Bi in 3+ oxidation states. The electrochemical evaluation of the synthesized catalysts revealed its excellent stability in the neutral and basic pH in the dark and under illumination however, the catalyst was unstable in the harsh acidic medium (pH = 2). A low resistance to electron transfer and better charge retention ability was witnessed by EIS and chronopotentiometry. The photocatalytic activity of the synthesized catalysts was evaluated in the exposure of complete spectrum and visible region of natural sunlight for the removal of chemically stable substrates such as 2-nitrophenol and 2-chlorophenol. The catalysts showed considerably high activity for the removal of both the substrates in the exposure of natural sunlight, whereas 25% less activity was witnessed in visible region in the same span of time. In addition to degradation, the catalyst also exhibited a substantial activity for mineralization (TOC removal). The catalysts unveiled reproducible activity in the repeated scans. The chemical stability and sustained activity both in the complete spectrum and visible region designate it as a potential candidate for photocatalytic environmental remediation.
Introduction
Although photocatalysis is an escalating area in water decontamination, the associated technical issues such as the non-availability of active narrow bandgap photocatalysts, the use of complicated experimental setup with artificial excitation sources hinder its wide spread commercial applicability. The use of technologically complicated and expensive excitation sources not only restricts this conclusive decontamination process to lab-scale but adds to its cost enormously. It is believed that the cheap and ever renewable sunlight can endorse the wide spread large-scale commercial applicability of photocatalysis by replacing the artificial light sources. The inability of the active photocatalysts like TiO2 (3.2 eV) and ZnO (3.0 eV), to harvest the maximal portion of the solar spectrum has forced the scientific community to investigate new sunlight responsive material with superior activity. In this context, besides the alteration of already existing single metal visible light active oxide semiconducting materials such as Bi2O3, WO3, V2O5, CeO2 etc., for better activity, the exploration of bimetallic oxide semiconductor materials as photocatalysts is an innovative approach. The co-existence of two chemically compatible metals, besides improving the structural properties, can strongly influence the optical and photocatalytic properties of bimetallic semiconductor oxides.1–17Iron bismuth oxide (FeBiO3) with a bandgap of 2.2 eV, chemical stability and optoelectronic properties is a versatile material for aqueous phase photocatalytic applications.18,19 The single phase FeBiO3 is ferroelectric and weakly ferromagnetic at room temperature.20,21 Although a number of synthetic routes are reported in the literature,22–30 however, its phase pure synthesis is still a topic of interest. It has been also reported that the synthesis of FeBiO3 by solid-state reaction at high temperature leads to residual impurities.31
The current study is an effort to develop phase pure sunlight responsive active photocatalysts. The bimetallic photocatalyst, FeBiO3, was synthesized by a user friendly approach using surfactant and the metallic ions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratios) by hydrogel route. The synthesized catalyst was characterized for optical, structural, electrochemical and morphological properties prior to photocatalytic experiments. The possible variations in the oxidation states of the individual metallic ions during the synthetic process was evaluated by XPS, whereas XRD and EDS were employed to verify the phase purity. Additionally, the phase purity was estimated by comparing the optical and structural properties with that of pure oxides. The stability of the synthesized catalyst was evaluated by cyclic voltammetry in the acidic and basic conditions in the dark and under illumination, whereas the resistance to the charge transfer and charge retention ability was estimated by EIS and chronopotentiometry, respectively. The photocatalytic activity of the synthesized catalyst was evaluated for the removal as well as mineralization of chemically stable and potential pollutants like 2-nitrophenol (2-NP) and 2-chlorophenol (2-CP). A number of chemical analysis tools such as HPLC, TOC, IC and GC-MS were employed to monitor the progress of degradation/estimation of intermediates, progress of mineralization, measurement of released ions and the identification of intermediates. The reusability of the synthesized catalysts was estimated by using the same catalyst for repeated scans.
:1 molar ratios) by hydrogel route. The synthesized catalyst was characterized for optical, structural, electrochemical and morphological properties prior to photocatalytic experiments. The possible variations in the oxidation states of the individual metallic ions during the synthetic process was evaluated by XPS, whereas XRD and EDS were employed to verify the phase purity. Additionally, the phase purity was estimated by comparing the optical and structural properties with that of pure oxides. The stability of the synthesized catalyst was evaluated by cyclic voltammetry in the acidic and basic conditions in the dark and under illumination, whereas the resistance to the charge transfer and charge retention ability was estimated by EIS and chronopotentiometry, respectively. The photocatalytic activity of the synthesized catalyst was evaluated for the removal as well as mineralization of chemically stable and potential pollutants like 2-nitrophenol (2-NP) and 2-chlorophenol (2-CP). A number of chemical analysis tools such as HPLC, TOC, IC and GC-MS were employed to monitor the progress of degradation/estimation of intermediates, progress of mineralization, measurement of released ions and the identification of intermediates. The reusability of the synthesized catalysts was estimated by using the same catalyst for repeated scans.
Experimental details
The bimetallic oxide, FeBiO3, was synthesized by hydrolysing the solution of equimolar ratio Fe3+ and Bi3+ ions with 0.5 M KOH (Sigma-Aldrich). The ferric nitrate (Fe(NO3)3·9H2O, Sigma-Aldrich) and bismuth nitrate (Bi(NO3)3·6H2O, Sigma-Aldrich) were used as the precursors for Fe3+ and Bi3+ ions. Prior to hydrolysis, the solution containing metallic ions were stirred with Triton X-100 (used as surfactant). In a typical synthesis the appropriate amount of above mentioned precursors of Fe3+ and Bi3+ ions containing 0.1 mole of the each ions were dissolved and stirred with 200 ml of 0.1% Triton X-100/deionized water/nitric acid mixture for 2 h. Prior to the addition of 0.5 M KOH, the solution was heated to 100 °C for half an hour. The solution containing the metal ions and surfactant was hydrolysed by the slow drop wise addition of 0.5 M KOH solution. The addition continued with stirring (1500 rpm) till pH 9. The hydrogel was heated at 200 °C until the formation of fine precipitates. The excessive KOH and surfactant was removed by successive addition of deionized water and decantation. At neutral pH, the precipitates were filtered and washed with 50:50 ethanol–acetone mixture to ensure the complete removal of surfactant. The collected precipitates were dried overnight, crushed and calcined in a muffle furnace at 500 °C for 4 h at a heating and cooling rate of 10 °C min−1. The α-Fe2O3, used in the study for the comparison, was synthesized by adopting the procedure detailed above, whereas Bi2O3 was pre-synthesized. The detailed synthesis of Bi2O3 is mentioned in our previous publications.12,13
For the comparison of optical properties such as solid-state absorption and diffuse reflectance spectra, the measurements were recorded by a Perkin Elmer UV-visible diffuse reflectance spectrophotometer (Lambda 650) in the wide range of 190–900 nm. The direct bandgaps of the materials were evaluated by transforming the reflectance data to F(R), the Kubelka–Munk function. The emission (PL), FTIR and Raman shift spectra of the materials were acquired by a fluorescence spectro-fluorophotometer, RF-5301 PC, Shimadzu, Japan at an excitation wavelength of 360 nm, FTIR spectrometer, Affinity 1, Shimadzu, Japan in 400 to 4000 cm−1 range using KBr pallet and a DXR Raman Microscope, Thermo Scientific, USA, equipped with 532 nm laser as the excitation source at 6 mW power, respectively. The XRD patterns of the pre and as-synthesized powders were recorded by Scintag XDS 2000 diffractometer, equipped with a Cu Kα radiation source, in 10 to 80° range. The XPS profile of synthesized FeBiO3 powder was assimilated in the binding energy range of 0 eV to 1100 eV by X-ray Photoelectron Spectrometer (PHI 5000 VersaProbe II, ULVAC-PHI Inc.). The oxidation states of Fe and Bi were estimated by applying curve fitting and comparing the data with standard values. The morphology of the synthesized powder was inspected by Field Emission Scanning Electron Microscope (FEI, Quanta FEG 450, Quorum Q150R ES, Quorum technologies Ltd. Ashford. Kent. England) at a voltage of 30 kV.
The electrochemical behavior and stability of the synthesized FeBiO3, in the dark and under illumination, was carried out by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and chronopotentiometry (CP). For CV, EIS and charge–discharge analysis, a VSP multi-channel potentiostat (Bio-logic Science Instrument, USA) equipped with Ec-lab software and a three electrode system namely; glassy carbon (GC), platinum and Ag/AgCl saturated electrodes as working, counter and reference electrodes, respectively. For the modification of GCE (working electrode), a sonicated dispersion of FeBiO3 in chloroform was coated at the surface. The fitting of EIS Nyquist plots was performed by Zfit (Ec-lab software, Bio-logic Science Instruments, USA). Except for CV analysis which was performed by using 0.1 M (pH 9) phosphate buffer, 0.1 M (pH 7) phosphate buffer and 0.1 M (pH 2) phosphate buffer electrolyte to evaluate the stability under varying pH environment, all the other measurements were executed in 0.1 M KCl solution. A 50 watt halogen lamp was used as a light source for the measurements under illumination.
The photocatalytic performance of FeBiO3 was assessed in complete spectrum and visible region of sunlight exposure for the degradation/mineralization of 2-NP and 2-CP by exposing the catalysts/phenolic substrate suspension. Pyrex® glass reactor was utilized as a UV cutoff filter. The dimensions and surface area of the glass reactor were 15.5 (diameter) × 2.5 (height) and 189 cm2, respectively. The optimization of the amount of catalyst was carried out for the removal of 2-NP. In a typical experiment, 150 ml of 2-NP (50 ppm) solution loaded with 10, 30, 70, 100, 200, 300 and 500 mg of the catalyst was exposed to sunlight for a period of 60 min. A linear increase in the degradation of 2-NP was observed till 100 mg of catalyst loading with no significant change afterwards. Based on the study, 100 mg/150 ml suspension was used in the proceeding studies. To establish the adsorption–desorption equilibrium between the catalysts and substrate, prior to exposure in sunlight, the suspension was kept in the dark for 60 min. During the sunlight exposure with an illumination of 1000 ± 100 × 102 lx, the samples were drawn at regular intervals and subjected to HPLC (HPLC, (SPD-20A, Shimadzu Corporation, Japan), TOC (TOC-VCPH total carbon analyzer, Shimadzu Corporation, Japan and IC (Thermo scientific, USA, ion chromatograph, Dionex (ICS-5000 + EG) Eluent Generator) analysis for the monitoring of degradation, mineralization and estimation of released ions during the photocatalytic process.32–36
Results and discussion
The comparison of the solid state absorption spectra α-Fe2O3, Bi2O3 and FeBiO3 in the broad spectral range of 200 to 900 nm is presented in Fig. 1(a), where compared to pure Bi2O3, a significantly strong absorption of FeBiO3 and α-Fe2O3, in the visible region (450–750 nm) is observable. Additionally, an inclination of the absorption pattern of FeBiO3 towards that of α-Fe2O3 was also witnessed however, instead of a sharp absorption edge, as observed for α-Fe2O3, a consistently increasing absorption trend was perceived. Moreover, FeBiO3 showed even better absorption in the visible region as compared to α-Fe2O3. The strong absorption in the visible region by α-Fe2O3 is attributed to the inter d–d transitions. In FeBiO3, as ∼50% of Bi3+ are replaced by Fe3+, its properties are strongly influenced by Fe3+. It seem quite obvious that the presence of Fe3+ entities in the structure and the existence of d–d transitions overshadows the absorption spectra and a comparable rather higher absorption of FeBiO3 was observable. The valence bands of the three powders i.e. α-Fe2O3, Bi2O3 and FeBiO3 are constituted by the O2− 2p orbitals, whereas the conduction bands are composed of Fe3+ (3d), Bi3+ (6p) and Fe3+Bi3+ (3d + 6p), respectively. These absorption edges represent the band gap excitations in the polymorphs. The graphical evaluation of the bandgap obtained by plotting (F(R) × hν)1/2 versus hν (photon energy) of Bi2O3 is presented in Fig. 1(b), whereas that of α-Fe2O3 and FeBiO3 are presented in the inset of Fig. 1(b). The evaluated bandgaps of 2.1 eV, 2.15 eV and 2.7 eV for α-Fe2O3, FeBiO3 and Bi2O3, respectively.37,38 A mild blue shift compared to the bandgap of α-Fe2O3 and a significant red shift compared to that of Bi2O3 demonstrates the leading role of Fe3+ states in establishing the band edge of FeBiO3. | ||
| Fig. 1 (a) The comparison of the solid-state absorption spectra of FeBiO3, α-Fe2O3 and Bi2O3 in 200–900 nm range, (b) the graphical evaluation of direct band edge position of Bi2O3, and the inset of (b) shows the same for FeBiO3 and α-Fe2O3. | ||
Fig. 2 shows the comparison of the normalized PL spectra of α-Fe2O3, FeBiO3 and Bi2O3 at an excitation wavelength of 350 nm and slit width of 5 nm. It is important to mention here that compared to FeBiO3, the emission intensities of α-Fe2O3 and Bi2O3, under identical measurement conditions were significantly higher. The characteristic broad bands were observed that depicted the wide range of transitions in the materials. The maxima's of the bands for α-Fe2O3, FeBiO3 and Bi2O3 at 580 nm, 570.5 nm and 469 nm corresponding to 2.07 eV, 2.21 eV and 2.63 eV, respectively were in accordance with the literature values.13,39 These emissions represented the Fe3+ 3d–O2− 2p, Fe3+Bi3+ (3d + 6p)–O2− 2p, and Bi3+ 6p–O2− 2p de-excitations (conduction to valence band) in α-Fe2O3, FeBiO3 and Bi2O3, respectively. The appearance of a distinct maxima at a position different than that of the parent oxides (α-Fe2O3 and Bi2O3) verified the phase purity of FeBiO3 and additionally, from material and photocatalysis point of view, verified the rearrangement of the conduction band composed of Fe3+ (3d) and Bi3+ (6p) states to a lower position thus lowering the bandgap of the material.
 | ||
| Fig. 2 The comparison of the normalized PL spectra of α-Fe2O3, FeBiO3, and Bi2O3 at an excitation wavelength of 325 nm and slit width of 5 nm. | ||
To ensure the mutual insertion of Fe3+ and Bi3+ in the crystal structure of FeBiO3 without the formation of other metal oxide offshoots, the FTIR analysis of Bi2O3, FeBiO3 and α-Fe2O3 was performed and compared in Fig. 3(a). Being rigid in nature, the information regarding the metal–oxygen–metal bond appear in the high energy region of 400–600 cm−1.40 The band at 541.99 cm−1 in the FTIR spectrum of Bi2O3 corresponded to the stretching vibrations of Bi3+–O–Bi3+ bond, whereas the corresponding vibrations of Fe3+–O–Bi3+ and Fe3+–O–Fe3+ bonds in FeBiO3 and α-Fe2O3 appeared at 553.57 cm−1 and 578.64 cm−1, respectively. The order of the vibrational energy for the corresponding bonds seems quite logical as the insertion of Fe3+ ions in the crystal structure, being lighter in nature, reduces the rigidity of the system compared to that of Bi2O3 thus vibrates at lower energy compared to that of Bi3+–O–Bi3+ bond. On the contrary, the addition of Bi3+, being heavier than Fe3+ in the structure restricts the magnitude of vibrations therefore shifted to higher energy compared to Fe3+–O–Fe3+ bond. The shifting in the position of the FTIR therefore validated the mutually shared oxygen positions by Fe3+ and Bi3+ ions in FeBiO3. The Raman spectroscopy being contemporary to FTIR is also regarded as a powerful tool in estimating the probable structural changes. The comparison of the Raman shift spectra of Bi2O3, FeBiO3 and α-Fe2O3 is presented in Fig. 3(b), where compared to that of Bi2O3 and α-Fe2O3, a significant shifting in the peak positions for FeBiO3 is observable. The major peaks representing the A1 modes for FeBiO3 appeared at 158.95 cm−1 and 490.64 cm−1, whereas the E modes were identified at 210.05 cm−1, 277.55 cm−1, 391.33 cm−1, 533.07 cm−1 and 588.03 cm−1. These values were in close agreement with that already mentioned in the literature with the discussion on their origin.41 The mismatch between the peaks of FeBiO3 with that of α-Fe2O3 and Bi2O3 as well as the shifts in the peak positions verified the co-existence of Fe3+ and Bi3+ in the structure.
 | ||
| Fig. 3 The comparison of the (a) FTIR and (b) normalized Raman spectra of Bi2O3, FeBiO3, and α-Fe2O3. | ||
Fig. 4 shows the comparison of the XRD patterns of Bi2O3, FeBiO3 and α-Fe2O3. The major reflections of α-Fe2O3 appeared at 2θ values of 24.14°, 33.14°, 35.65°, 40.86°, 49.57°, 54.15°, 62.40° and 64.04°, whereas the reflections of appreciable intensity for Bi2O3 appeared at 25.85°, 27.45°, 28.15°, 32.2°, 33.25°, 35.20°, 37.15°, 42.35°, 46.30°, 52.37° and 61.88°. The XRD pattern of α-Fe2O3 corresponded to rhombohedral geometry with the crystallite size of ∼40.2 nm matched with JCPDS # 33-0664. A monoclinic geometry (JCPDS # 41-1449) was assessed for Bi2O3, whereas a crystallite size of ∼36.2 nm was calculated by using the parameters of the most intense reflection at 27.45°. The prominent reflections of FeBiO3 were experiential at the 2θ values of 22.45°, 31.95°, 39.55°, 45.55°, 51.71°, 57.10°, 67.25°, 71.17° and 76.09° that corresponded to (101), (110), (021), (202), (113), (300), (220), (303) and (312) indices of rhombohedral structure (JCPDS # 20-0169). An average crystallite size of ∼19.2 nm was calculated using the intensity, d-spacing and FWHM values of the intense reflections. The intensity and sharpness of the peaks reflected the well-defined crystal structure of FeBiO3 with high crystallinity.
 | ||
| Fig. 4 The comparison of the XRD patterns of Bi2O3, FeBiO3 and α-Fe2O3. The dotted lines shows the major reflections of FeBiO3. | ||
The XPS wide angle survey scan of as-synthesized FeBiO3 is presented in Fig. 5(a), where the peaks against the corresponding binding energies of the core and splitted levels of Bi, Fe and O are observable. The C 1s peak was observed at 285.02 eV. The estimation of the probable variations in the oxidation states of the individual components during the synthesis process, especially Fe and Bi, was performed by the identification of the submerged peaks by deconvolution and curve fitting. The raw and fitted peaks for Bi 4f splitted levels are presented in Fig. 5(b). The deconvolution of the Bi 4f located the additional low intensity peaks at 157.5 eV (4f7/2) and 162.8 eV (4f5/2) besides the intense peaks at 159.5 eV (4f7/2) and 164.7 eV (4f5/2). Such type of behavior is not reported in the literature as the existence of higher oxidation states such as Bi5+ leads to the positive increment in the binding energy.42 Therefore, based on the evaluation of all the possibilities, it was established that the change in the chemical environment resulted in the shifting of the Bi3+ peaks to lower energy. The splitted peaks of 4f7/2 and 4f5/2 peaks due to Bi3+–O–Bi3+ linkage appeared at 159.5 eV and 164.7 eV, respectively, whereas the Fe3+–O–Bi3+ shifted the same to lower energy values of 157.5 eV and 162.8 eV, respectively. The fitted Fe 2p splitted levels are presented in Fig. 5(c). The peaks of Fe 2p1/2 and Fe 2p3/2 levels at 726.23 eV and 712.35 eV, respectively, verified the presence of Fe in 3+ oxidation state. Additionally, the presence of Fe3+ characteristic satellite at 718.7 eV and the absence of additional peaks further authenticated the findings. As presented in Fig. 5(d), the fitting of O 1s spectra resolved in three peaks of variable intensities. The peak at ∼532.3 eV characterize the chemisorbed surface hydroxyl groups, whereas the main peak at ∼530.28 eV represent the skeletal oxygen, whereas the peak due to the singly charged surface O-groups was observed at low energy position of ∼528.52 eV.
 | ||
| Fig. 5 The XPS analysis of FeBiO3 (a) survey scan (b) Bi 4f splitted core levels (c) Fe 2p splitted core levels and (d) O 1s splitted levels. | ||
The comparison of the FESEM images of as-synthesized FeBiO3 at 100000×, 200000×, 300000× and 400000× are presented in Fig. 6(a–d), respectively. A narrow particle size distribution with the uniformity in the shape was observable that validated the applicability of the used synthetic route and significance of surfactant in controlling the morphology along with other physical factors such as temperature, rate of KOH addition etc. The evaluated crystallite size of ∼19.2 nm was also validated by the FESEM analysis. It was also noticed that the larger particles (>40 nm) were the aggregates of the smaller ones. In the high resolution images (Fig. 6(d)), the particles with spherical shapes are also noticeable. Fig. 7 shows the location of the targeted or focused point for the EDS elemental analysis of the synthesized FeBiO3, whereas the EDS spectra and the elemental composition of the powder is presented in the inset. The analysis revealed the presence of Fe and Bi in 1:1 and also verified the phase purity of the material.
 | ||
| Fig. 6 The comparison of the FESEM images of FeBiO3 at various resolutions (a) 100000× (b) 200000× (c) 300000× and (d) 400000×. | ||
 | ||
| Fig. 7 The EDS analysis of the synthesized FeBiO3. The inset shows the EDS spectra and the elemental composition of the components of the sample. | ||
The stability of the synthesized FeBiO3 was examined under various pH conditions i.e. acidic (pH = 2), neutral (pH = 7.0) and basic (pH = 9.0) by cyclic voltammetry (CV) in the dark and under illumination and the comparison is presented in Fig. 8(a) and (b). It is important to mention here that two scans were recorded both in the dark and under illumination however, the results of the Ist cycle in the dark and under illumination are compared. In 0.1 M pH 2 phosphate buffer electrolyte, two distinct reduction peaks appeared at −0.19 V vs. RHE (−0.51 V vs. Ag/AgCl) and −0.57 V vs. RHE (−0.89 V vs. Ag/AgCl) in the forward cathodic direction both in the dark and under illumination that indicated the dissolution or surface erosion of the powder supported on GCE. The Ist peak at −0.19 V vs. RHE (−0.51 V vs. Ag/AgCl) was attributed to the electro-reduction of surface Bi3+–O− type species in the acidic environment leading to the formation of BiO2− species, whereas the IInd peak was assigned to the further reduction of dissolved species BiO2− and possible bulk deposition of released Bi. The results similar to our findings are already reported in the literature.43–45 In the backward anodic direction, an oxidation peak due to the oxidation of Fe2+ into Fe3+ was appeared at +0.29 V vs. RHE (−0.03 V vs. Ag/AgCl). The Fe2+ ion formed in the forward cathodic scan from the reduction of surface Fe3+ however, the broad reduction peak of Bi3+–O− species obscured the reduction peak of Fe3+. In the IInd cycle, probably due to the deposition of released Bi at the electrode surface, the electrode surface was passivated, therefore no reduction peaks were observed in the dark. Additionally, the anodic and cathodic peak currents were substantially decreased. A similar behavior was observed under illumination, however, the reduction peak currents were significantly enhanced as compared to that of dark conditions.
 | ||
| Fig. 8 The electrochemical evaluation of FeBiO3 in the dark and under illumination (a & b) The CV's at various pH values (c) EIS Nyquist plot of FeBiO3 in dark and under illumination (d & e) exploded view of high and low frequency regions of EIS spectra (f) charge–discharge profile of FeBiO3 at neutral pH. | ||
Contrary to acidic medium (pH = 2), in the neutral 0.1 M pH 7 phosphate buffer, the susceptibility of FeBiO3, both in the dark and under illumination, towards reduction was dramatically reduced and a drastic decrease in the cathodic peak intensities elaborated its better stability in the neutral medium. However, both in the dark and under illumination, in the anodic region, two low intensity broad oxidation peaks were noticed at +0.11 V vs. RHE (−0.5 V vs. Ag/AgCl) and +0.45 V vs. RHE (−0.16 V vs. Ag/AgCl). As no significant change in the peak intensity was noticed under illumination, the formation of the respective metallic hydroxides under external bias was identified as the source in this regard. In the IInd cycle, the reduction of dissolved Bi(OH) was appeared at −0.28 V vs. RHE (−0.89 V vs. Ag/AgCl) in the dark. The decreased intensity of the oxidation peaks and the absence of the reduction peak under illumination further strengthen the view of the stability of FeBiO3 under illumination.
In basic medium (0.1 M pH 9 phosphate buffer), the background current at positive potential was significantly higher that suppressed the reduction peak current intensities considerably, therefore, the CV's were recorded in the potential range of +0.73 V vs. RHE to −0.27 V vs. RHE (0 V vs. Ag/AgCl to −1.0 V vs. Ag/AgCl). In the forward cathodic scan, three barely detectable reduction peaks were emerged at +0.23 V vs. RHE (−0.5 V vs. Ag/AgCl) due to reduction of bismuth oxide, at +0.06 V vs. RHE (−0.67 V vs. Ag/AgCl) for the reduction of Fe3+ and at −0.21 V vs. RHE (−0.94 V vs. Ag/AgCl) for the deposition of released bismuth. No clear oxidation peak appeared in the backward anodic direction. The disappearance of reduction peaks in the 2nd cycle confirmed the deposition of the released Bi at the electrode surface. Compared to dark, the higher peak currents intensities under illumination, were attributed to the increased conductivity of the films. The analysis of the CV patterns revealed the adequate stability in neutral and basic pH, whereas FeBiO3 encountered dissolution of both metallic components (Fe and Bi) in harsh conditions of acidic pH.
The comparison of the alternating current impedance measurements of FeBiO3/GCE, in the dark and under illumination, is presented in Fig. 8(c). A measurable decrease in the charge transfer resistance of as-synthesized FeBiO3 was observable both in low and high frequency range as shown in Fig. 8(d) and (e) that depicted the increased conductivity or better charge transfer ability under illumination. In terms of photocatalytic activity, this observation may be correlated to the expected better mobility of charges in the material under illumination.17,33 The charge–discharge behavior of FeBiO3/GCE was evaluated at 0.1 A m−2 current density. The enhanced magnitude of the discharging curve, under illumination demonstrated the better charge retaining ability of the material. The charge discharge profiles are shown in Fig. 8(f).
The photocatalytic performance of synthesized FeBiO3 was evaluated for the removal (degradation/mineralization) of 2-NP and 2-CP in complete spectrum and visible region sunlight exposure. The adsorption of the phenolic substrates on as-synthesized FeBiO3 was estimated by retaining the catalyst/substrate suspension in the dark for 1 h before the exposure. The FeBiO3 exhibited significantly high adsorption of ∼21% and ∼17% of 2-NP and 2-CP, respectively. The comparison of the percentage degradation of 2-NP, extracted from the decrease in peak height of 2-NP in HPLC chromatograms, as a function of sunlight (complete spectrum/visible region) is presented in Fig. 9(a). In the initial 30 min of complete spectrum sunlight exposure, ∼46% of 2-NP was removed whereas ∼15% was removed in visible region of sunlight exposure for the same period. Although ∼94% of 2-NP was removed in 120 min, however the complete removal was witnessed in 240 min of complete spectrum sunlight exposure. Probably the scarce access of ROS to the 2-NP substrate molecules due to the decreased concentration is the possible reason for this effect. In 240 min of visible region exposure, ∼80% of the substrate was degraded. Although the degradation process was slow in the visible region, however a consistent decrease in the concentration of 2-NP was noticed with the increasing exposure time. The rate constants (k) for the removal of 2-NP under both the conditions of exposure were estimated by applying the Langmuir–Hinshelwood kinetic model for pseudo first order reactions. The evaluation extracted by plotting ln(Co/C) versus the sunlight exposure time is presented in Fig. 9(b). The kinetic model was validated under both the experimental conditions and the rate constants of 0.023 min−1 and 0.0059 min−1 for the removal in complete spectrum and visible sunlight exposure, respectively. The significant difference in the rate of removal of 2-NP may be attributed to the contribution of the ≤5% UV portion in the complete spectrum sunlight. The other plausible is the blocking of the incident visible photons by 2-NP that has extended absorption band in the visible as well as UV region.
 | ||
| Fig. 9 The comparison of the percentage degradation and rates of degradation of 2-NP (a and b) and 2-CP (c and d) in the exposure of complete spectrum and visible region of sunlight in the presence of FeBiO3. | ||
The comparison of the time-scale percentage degradation of 2-CP in the complete spectrum and visible region exposure of sunlight is presented in Fig. 9(c). The degradation of 2-CP in the initial 30 min of the exposure in complete spectrum sunlight was comparable to that of 2-NP, whereas marginally higher i.e. ∼19% compared to ∼15% for 2-NP, in the visible region exposure. With the passage of time, a faster degradation of 2-CP, compared to 2-NP, under the both exposure conditions, was noticed. The probable reason for this effect is the absorption of photons by the 2-NP substrate that is extends from the visible region to UV region, whereas 2-CP has the weak absorption in the UV region below 300 nm. The absorption of photons by the substrate (2-NP) reduces the accessibility of the photons to the catalyst particles. After 240 min of exposure ∼100% and ∼85% of 2-CP was degraded in complete spectrum and visible region sunlight exposure, respectively. The graphical evaluation of the rate constants (k) is presented in Fig. 9(d). The evaluated rate constants for the degradation of 2-CP was 0.025 min−1 and 0.0071 min−1. Additionally, the Langmuir–Hinshelwood kinetic model was also validated with acceptable correlation.
The comparison of the mineralization (TOC removal) of 2-NP and 2-CP in the complete spectrum and visible region sunlight exposure is presented in Fig. 10(a). The synthesized FeBiO3 efficiently mineralized both the substrates in the complete spectrum sunlight exposure and reduced the initial TOC contents of 15.6 ppm and 16.8 ppm for 30 ppm of 2-NP and 2-CP to 0.62 ppm and 0.25 ppm, respectively, in 240 min of exposure. The graphical evaluation of rate constants for TOC removal of 2-NP and 2-CP in the exposure of complete spectrum and visible region of sunlight is presented in Fig. 10(b). The markedly lower rate of mineralization compared to that of degradation clearly reflected multistep nature of mineralization as compared to degradation process. Additionally, the rapid removal of the substrates also proposes the ease of degradation as compared to mineralization that is accomplished by any major or minor structural change in the substrate changing its identity as a new compound. Therefore, the degradation does not essentially mean the removal rather it is the conversion of substrate into new entity. The time-scale HPLC profiles of all the samples and GCMS analysis of the selected sample confirmed the formation of intermediates during the degradation process. The intermediates are further interacted by the oxidants leading to the formation of smaller fragments and later conversion to CO2 in successive interactions. The pictorial representation of the above discussion is presented in Scheme 1.
 | ||
| Fig. 10 The comparison of (a) TOC removal (ppm) and (b) graphical evaluation of rate constants for TOC removal of 2-NP and 2-CP in the exposure of complete spectrum and visible region of sunlight in the presence of FeBiO3. | ||
 | ||
| Scheme 1 The plausible mechanism of 2-CP and 2-NP degradation over FeBiO3 in sunlight exposure. | ||
The fate of the released ions during the degradation/mineralization process was monitored by ion chromatography (IC). Contrary to the expected Cl− and NO2− ions in the removal of 2-CP and 2-NP, respectively, ClO3− and NO3− ions were observed under the subjected instrumental conditions. The presence of these ions was validated by successive analysis of the standards. As estimated by the comparison of the peak heights of the standards and samples, the time-scale profiles of ClO3− and NO3− ions in complete spectrum and visible region illumination of sunlight are presented in Fig. 11(a). Interestingly, the concentration of these ions was significantly lower than the expected theoretical values. This situation led to the conclusion that the Cl− and NO2− ions are further interacted by either the photon generated ROS or the reactive sites such as holes (h+) and being transformed to the respective gases, Cl2 and NO2, that escape from the reactor during the course of degradation process. It has been proposed that NO2− ions undergo photolysis to NO˙2 in the aqueous system with the generation of hydroxyl radicals. The plausible routs are proposed as below.46
| NO2− + H+ + hν → NO˙2 + HO˙ | (1) |
| NO2− + HO˙ → NO˙2 + HO− | (2) |
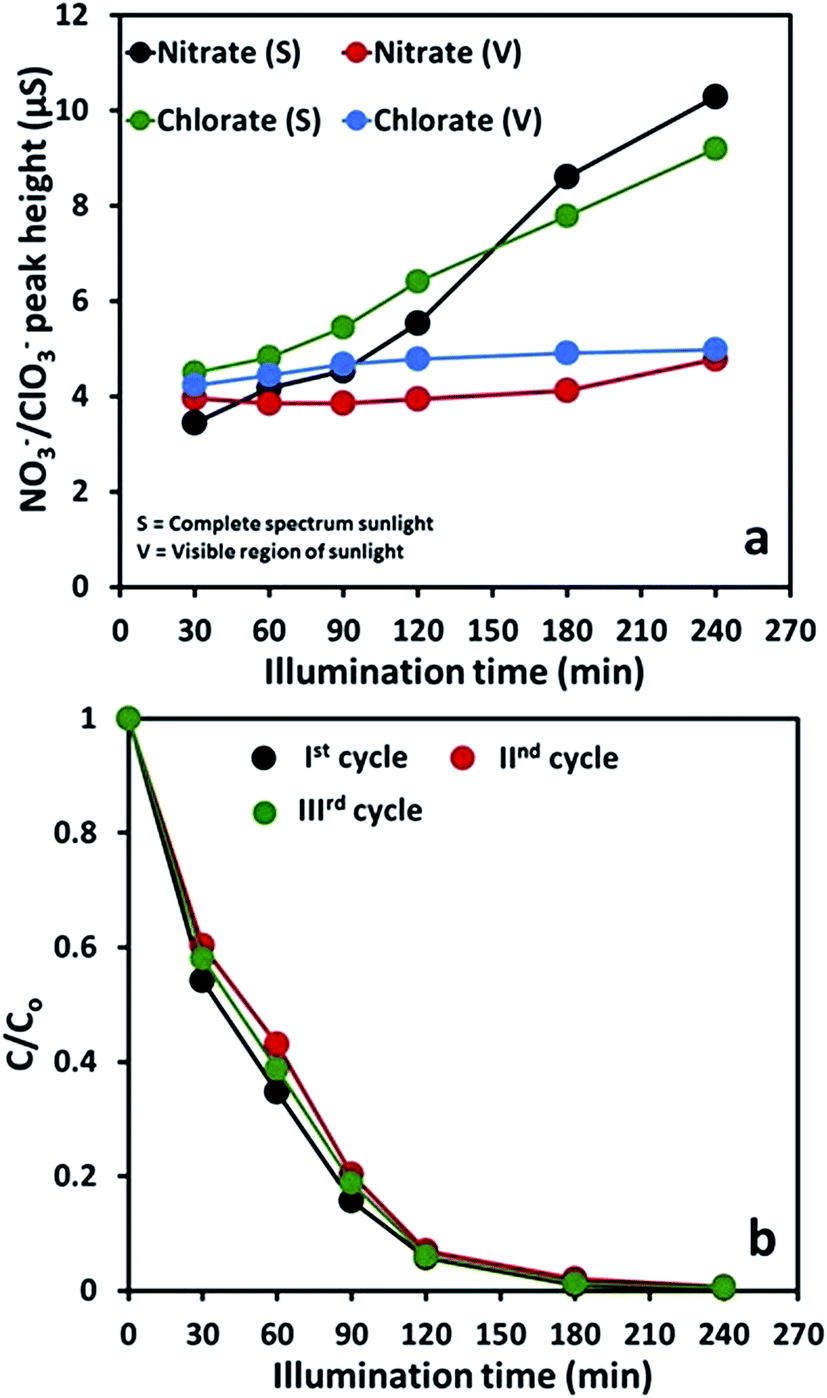 | ||
| Fig. 11 (a) The comparison of the major released ions during the degradation of 2-NP and 2-CP in the exposure of complete spectrum and visible region of sunlight in the presence of FeBiO3. (b) Evaluation of the reusability of FeBiO3 for the degradation of 2-NP in complete spectrum sunlight exposure. | ||
The proposed mechanism indicate that the nitrite ions simultaneously act as producers and consumers of hydroxyl radicals under illumination. The above mechanisms seems equally applicable on the photocatalytic systems as all the species involved are either already present in the system or generated under illumination. Another possibility that we have already discussed in our previous communication,5 is the direct donation of electrons by the nitrite ions to the photon generated holes at the surface of photocatalyst.
| NO2− + h+ → NO2↑ | (3) |
The direct evidence for the escape of the NO2 gas during the degradation process was the formation of brownish-yellow vapors. It is important to mention here that in some of our studies with ZnO, we observed the presence of nitrite ions in substantial concentration.40 Based on eqn (1), it was presumed that the presence of H+ ions in the solution is vital for the conversion of nitrite ions into NO2 gas. This hypothesis was verified by measuring the pH during the course of reaction which revealed that the pH of the system persisted in the acidic range i.e. 5.5 to 6.5. These observations validated the proposed mechanism in eqn (1). The same analogy is equally probable for the conversion of chloride ions into either Cl2 or HCl gas in the acidic pH conditions.
The reusability of the synthesized catalyst was evaluated for the removal of 2-NP in the complete spectrum sunlight exposure. The comparison of the three successive scans performed with the same catalyst with fresh 30 ppm 2-NP solution is presented in Fig. 11(b). The catalyst showed acceptable sustained performance and no significant decrease in the activity in the repeated scans.
Conclusions
The study revealed the suitability of the adopted procedure for the phase pure and morphology controlled synthesis of FeBiO3. The presence of both Fe and Bi in the crystal structure of FeBiO3 imparted better structural, electrochemical and optical properties that classified it as a useful addition for sunlight photocatalytic decontamination applications. The catalyst demonstrated significantly high activity for the removal (degradation as well as mineralization), of 2-NP and 2-CP both in complete spectrum and visible sunlight exposure. The ions released as a consequence of mineralization are further interacted by photon generated reactive oxygen species and released as respective gases.Acknowledgements
Iqbal M. I. Ismail, A. Hameed and M. Aslam are thankful to CEES, KAU and Ministry of Higher Education, KSA, for support.References
- D. Bahnemann, Sol. Energy, 2004, 77, 445 CrossRef CAS.
- W. Bahnemann, M. Muneer and M. M. Haque, Catal. Today, 2007, 124, 133 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1 CrossRef CAS.
- M. R. Hoffmann, S. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- M. Aslam, I. M. I. Ismail, S. Chandrasekaran and A. Hameed, J. Hazard. Mater., 2014, 276, 120 CrossRef CAS PubMed.
- M. Kositzi, I. Poulios, S. Malato, J. Caceres and A. Campos, Water Res., 2004, 38, 1147 CrossRef CAS PubMed.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1 CrossRef CAS.
- A. Mills and S. le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1 CrossRef CAS.
- M. Aslam, I. M. I. Ismail, N. Salah, S. Chandrasekaran, M. T. Qamar and A. Hameed, J. Hazard. Mater., 2015, 286, 127 CrossRef CAS PubMed.
- S. K. Pardeshi and A. B. Patil, Sol. Energy, 2008, 82, 700 CrossRef CAS.
- Photocatalysis: Fundamentals and Applications, ed. E. Pelizzetti and N. Serpone, Wiley, New York, USA, 1989 Search PubMed.
- A. Hameed, T. Montini, V. Gombac and P. Fornasiero, J. Am. Chem. Soc., 2008, 130, 9658 CrossRef CAS PubMed.
- A. Hameed, M. Aslam, I. M. I. Ismail, N. Salah and P. Fornasiero, Appl. Catal., B, 2015, 163, 444 CrossRef CAS.
- P. Pichat, Photocatalysis and Water Purification: From Fundamentals to Recent Applications, Wiley-VCH Verlag, Weinheim, Germany, 1st edn, 2013 Search PubMed.
- S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, J. Hazard. Mater., 2009, 170, 560 CrossRef CAS PubMed.
- M. Schiavello, Photocatalysis and Environment: Trends and Applications, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1988 Search PubMed.
- M. Aslam, M. T. Qamar, M. T. Soomro, I. M. I. Ismail, N. Salah, T. Almeelbi, M. A. Gondal and A. Hameed, Appl. Catal., B, 2016, 180, 391 CrossRef CAS.
- T. J. Park, G. C. Papaefthymiou, A. J. Viescas, A. R. Moodenbough and S. S. Wong, Nano Lett., 2007, 7, 766 CrossRef CAS PubMed.
- S. M. Selbach, T. Tybell, M. A. Einarsrud and T. Grande, Chem. Mater., 2007, 19, 6478 CrossRef CAS.
- G. Catalan and J. F. Scott, Adv. Mater., 2009, 21, 2463 CrossRef CAS.
- B. Liu, B. Hu and Z. Du, Chem. Commun., 2011, 47, 8166 RSC.
- S. Li, Y. H. Lin, B. P. Zhang, Y. Wang and C. W. Nan, J. Phys. Chem. C, 2010, 114, 2903 CAS.
- T. Choi, S. Lee, Y. Choi, V. Kiryukhin and S. W. Cheong, Science, 2009, 324, 63 CrossRef CAS PubMed.
- C. Chen, J. R. Cheng, S. W. Yu, L. J. Che and Z. Y. Meng, J. Cryst. Growth, 2006, 291, 135 CrossRef CAS.
- D.-C. Jia, J.-H. Xu, H. Ke, W. Wang and Y. Zhou, J. Eur. Ceram. Soc., 2009, 29, 3099 CrossRef CAS.
- J. Luo and P. A. Maggard, Adv. Mater., 2006, 18, 514 CrossRef CAS.
- U. A. Joshi, J. S. Jang, P. H. Borse and J. S. Lee, Appl. Phys. Lett., 2008, 92, 242106 CrossRef.
- F. Gao, Y. Yuan, K. F. Wang, X. Y. Chen, F. Chen, J.-M. Liu and Z. F. Ren, Appl. Phys. Lett., 2006, 89, 102506 CrossRef.
- F. Gao, X. Chen, K. Yin, S. Dong, Z. Ren, F. Yuan, T. Yu, Z. Zou and J. M. Liu, Adv. Mater., 2007, 19, 2889 CrossRef CAS.
- X. Y. Chen, T. Yu, F. Gao, H. T. Zhang, L. F. Liu, Y. M. Wang, Z. S. Li and Z. G. Zou, Appl. Phys. Lett., 2007, 91, 022114 CrossRef.
- T. Xian, H. Yang, J. F. Dai, Z. Q. Wei, J. Y. Ma and W. J. Feng, Mater. Lett., 2011, 65, 1573 CrossRef CAS.
- M. T. Qamar, M. Aslam, I. M. I. Ismail, N. Salah and A. Hameed, ACS Appl. Mater. Interfaces, 2015, 7, 8757 CAS.
- M. Aslam, M. T. Soomro, I. M. I. Ismail, N. Salah, M. W. Ashraf, H. A. Qari and A. Hameed, Arabian J. Chem., 2015 DOI:10.1016/j.arabjc.2015.05.001.
- M. Aslam, I. M. I. Ismail, S. Chandrasekaran, T. Almeelbi and A. Hameed, RSC Adv., 2014, 4, 49347 RSC.
- M. Aslam, I. M. I. Ismail, S. Chandrasekaran, H. A. Qari and A. Hameed, Water, Air, Soil Pollut., 2015, 226, 70 CrossRef.
- M. T. Qamar, M. Aslam, I. M. I. Ismail, N. Salah and A. Hameed, Chem. Eng. J., 2016, 283, 656 CrossRef CAS.
- J.-P. Xu, R.-J. Zhang, Z.-H. Chen, Z.-Y. Wang, F. Zhang, X. Yu, A.-Q. Jiang, Y.-X. Zheng, S.-Y. Wang and L.-Y. Chen, Nanoscale Res. Lett., 2014, 9, 188 CrossRef PubMed.
- Z. K. Liu, Y. J. Qi and C. J. Lu, J. Mater. Sci.: Mater. Electron., 2010, 21, 380 CrossRef CAS.
- T. Xian, H. Yang, J. F. Dai, Z. Q. Wei, J. Y. Ma and W. J. Feng, Mater. Lett., 2011, 65, 1573 CrossRef CAS.
- A. Hameed, M. Aslam, I. M. I. Ismail, S. Chandrasekaran, M. Kadi and M. A. Gondal, Appl. Catal., B, 2014, 160–161, 227 CrossRef CAS.
- D. Kothari, V. R. Reddy, V. G. Sathe, A. Gupta, A. Banerjee and A. M. Awasthi, J. Magn. Magn. Mater., 2008, 320, 548 CrossRef CAS.
- M. Ciszewski, A. Mianowski, P. Szatkowski, G. Nawrat and J. Adamek, Ionics, 2015, 21, 557 CrossRef CAS.
- V. Vivier, A. Régis, G. Sagon, J. Y. Nedelec, L. T. Yu and C. Cachet-Vivier, Electrochim. Acta, 2001, 46, 907 CrossRef CAS.
- S. M. Skogvold, Ø. Mikkelsen and K. H. Schrøder, Electroanalysis, 2005, 17, 1938 CrossRef CAS.
- R. Pauliukaitė, S. B. Hočevar, B. Ogorevc and J. Wang, Electroanalysis, 2004, 16, 719 CrossRef.
- D. Vione, V. Maurino, C. Minero and E. Pelizzetti, Chemosphere, 2004, 55, 941 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |