
Expedient, catalyst-free, three-component synthesis of fused tetrahydropyridines in water†
Perumal
Vinoth
a,
P. S. Ram
Prasad
a,
Thavaraj
Vivekanand
a,
C. Uma
Maheswari
a,
Subbiah
Nagarajan
a,
J. Carlos
Menéndez
b and
Vellaisamy
Sridharan
*a
aOrganic Synthesis Group, Department of Chemistry, School of Chemical and Biotechnology, SASTRA University, Thanjavur 613401, Tamil Nadu, India. E-mail: vsridharan@scbt.sastra.edu; vesridharan@gmail.com
bDepartamento de Química Orgánica y Farmacéutica, Facultad de Farmacia, Universidad Complutense, 28040 Madrid, Spain
First published on 22nd September 2015
Abstract
A catalyst-free, three-component reaction between amino alcohols, 1,3-dicarbonyl compounds and α,β-unsaturated aldehydes was developed for the synthesis of fused tetrahydropyridines in water. The β-enaminone formation-initiated domino sequence afforded oxazolo[3,2-a]pyridines and pyrido[2,1-b][1,3]oxazines diastereoselectivity in good yields involving Michael addition, intramolecular cyclization and iminium ion cyclization steps. This environmentally benign protocol is highly atom-economical where the only side product was two molecules of water and no catalyst or reagent was employed. Besides, in a single operation, four new bonds including two C–N, one C–O and one C–C bonds and two heterocyclic rings were created. The reaction was also effective in various green solvents such as glycerol, PEG-200 and lactic acid.
Introduction
Reactions that create several bonds in a single operation to generate complex molecules are remarkably important in contemporary organic synthesis. Among the several families of reactions discovered, multicomponent reactions hold a prominent place in generating diverse products in one-pot.1 These multicomponent reactions contribute a great extent to green chemistry owing to atom- and step-economy, waste reduction and high overall yields. Moreover, the number of steps of a synthetic sequence is significantly reduced and the purification of the intermediates is completely avoided in these reactions. The primary multicomponent reactions including Ugi, Hantzsch, Beginelli, Passerini, Povarov and Strecker reactions were generally found to be efficient to access biologically significant molecules including a large number of heterocyclic compounds. For instance, the simple and most common nitrogen heterocycles present in drug molecules, natural products and biologically active compounds including pyrroles, pyridines, and their benz-fused analogs such as indoles and quinolines were conveniently synthesized by means of multicomponent reactions.2Pyridines and their partially hydrogenated analogs including dihydro- and tetrahydropyridines are ubiquitous heterocyclic fragments of natural products and bioactive compounds. A large number of natural compounds exemplified by vitamin B6, nicotine, and commercial drug molecules actos, nifedipine and felodipine contain this scaffold. Furthermore, these analogs exhibit numerous interesting biological activities such as anti-inflammatory,3 antidepressant,4 anti-HIV,5 anticonvulsant,6 antiasthmatic,7 calcium channel blocking activity8 and many others. Consequently the development of synthetic methods to access pyridines and their partially hydrogenated derivatives is an essential goal in organic synthesis.9 Especially, 1,2,3,4-tetrahdydropyridines are significant owing to their unique pharmacological activities and their application in food chemistry.10,11
Tetrahydropyridines fused with other heterocyclic fragments such as pyrido[2,1-b][1,3]oxazines and oxazolo[3,2-a]pyridines are known to show versatile pharmacological properties. For instance, pyrido[2,1-b][1,3]oxazines bear anti-inflammatory, spasmolytic and antihypertensive activities,12 and oxazolo[3,2-a]pyridines displayed significant reversion of multi-drug resistance in Leishmania.13 In addition, oxazolo[3,2-a]pyridines are efficient antihypertensive14 and serve as excellent precursors to access chiral piperidine derivatives and alkaloids.15 Despite their significant bioactivity profile, methods allowing direct access to these fused tetrahydropyridines remained scarce.16,17 Generally, oxazolo[3,2-a]pyridines had been achieved by the reaction between enamines and amino alcohols under reflux condition albeit in lower yields.18 Recently, Wan and co-workers have reported a three-component procedure starting from enals, electron-deficient alkynes and hydroxyl-functionalized amines under acidic conditions.19 Furthermore, a two-step, Michael addition-condensation sequence have also been developed for the synthesis of oxazolo[3,2-a]pyridines.20 Most of these two-component protocols suffer with the difficulties associated with the isolation and purification of the acid-sensitive and unstable enamines and low yields in addition to the restriction in the substitution patterns.
Results and discussion
In view of the increasing importance of oxazolo[3,2-a]pyridines and pyrido[2,1-b][1,3]oxazines, we envisioned to develop a simple, three-component procedure starting from readily available 1,3-dicarbonyl compounds, cinnamaldehyde derivatives and amino alcohols in the absence of any catalyst to access these fused tetrahydroquinolines in a single operation. We have previously reported a four-component synthesis of 1,4,5,6-tetrahydropyridines in the presence of cerium(IV) ammonium nitrate catalyst21 and the application of this protocol was extended to access a number of heterocyclic systems.22 Herein, we further explore this methodology to access oxazolo[3,2-a]pyridines and pyrido[2,1-b][1,3]oxazines in water with the use of no catalyst. Lhommet and co-workers have also reported a related procedure restricted only to acrolein and with low yields.23 The combination of multicomponent technique, use of water or a green solvent as the reaction medium and involvement of no reagent or catalyst would be the ideal strategy to develop perfect green synthetic procedure that would address most of the twelve principles of green chemistry.24Our study commenced with a three-component reaction between ethanolamine 1a, ethyl acetoacetate 2a and cinnamaldehyde 3a in the presence of 5 mol% of CAN25 in water and the results are summarized in Table 1. Expectedly, the reaction proceeded smoothly to afford product 4a as a single diastereomer in 69% yield at 25 °C in a span of two hours reaction time, and change of catalyst to InCl3 did not improve the yield significantly (entries 1 and 2). To our delight, the catalyst-free condition was superior in water to the previous reactions furnishing a maximum yield of 75% (entry 3). With an aim to improve the yield, in the absence of any catalyst, we tuned the reaction condition by increasing the reaction time and temperature (entries 4–6). Nonetheless, no significant improvement in yield was noticed although the reaction was completed in one hour at 80 °C. Other modifications including the increase of the quantity of the reaction medium or use of 1.5 equivalents of amino alcohol 1a were failed to improve the yield further. In fact, use of large amount of water as reaction medium suppressed the yield to 66% (entries 7 and 8). Screening of other green solvents including glycerol, PEG-200 and lactic acid were also effective to provide the product. In all the three solvents, the reaction was completed in two hours, however, with almost identical yields of the previous conditions (entries 9–11). In a screen of solvents, we investigated a number of common organic solvents such as ethanol, acetonitrile, THF, DCM, DCE, toluene and dioxane (entries 12–18). Although the product was obtained in all the tested solvents, acetonitrile, DCM and DCE were superior affording around 80% of the product without any catalyst. Finally, we retained water as the reaction medium for its environmentally benign nature and other well-known benefits. No significant change in yield was observed when 3-propanolamine was used as the substrate under optimized reaction conditions (entries 19 and 20).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cpd | Reaction medium | Catalyst (5 mol%) | Temp. (°C) | Reaction time (h) | Yield (%) |
| a Reaction conditions: unless otherwise noted, all reactions were carried out with 1a/1b (1.3 mmol), 2a (1 mmol) and 3a (1 mmol) in 1 mL reaction medium. b Isolated yield. c 5 mL of water was used. d 1.5 equiv. of 1a was used. e Isolated enamine was used for the reaction. | ||||||
| 1 | 4a | H2O | CAN | 25 | 2 | 69 |
| 2 | 4a | H2O | InCl3 | 25 | 2 | 71 |
| 3 | 4a | H 2 O | — | 25 | 2 | 75 |
| 4 | 4a | H2O | — | 25 | 6 | 76 |
| 5 | 4a | H2O | — | 50 | 2 | 78 |
| 6 | 4a | H2O | — | 80 | 1 | 73 |
| 7 | 4a | H2Oc | — | 25 | 2 | 66 |
| 8 | 4a | H2Od | — | 25 | 2 | 76 |
| 9 | 4a | Glycerol | — | 25 | 2 | 70 |
| 10 | 4a | PEG-200 | — | 25 | 2 | 73 |
| 11 | 4a | Lactic acid | — | 25 | 2 | 71 |
| 12 | 4a | EtOH | — | 25 | 2 | 74 |
| 13 | 4a | MeCN | — | 25 | 2 | 78 |
| 14 | 4a | THF | — | 25 | 2 | 73 |
| 15 | 4a | DCM | — | 25 | 5 | 79 |
| 16 | 4a | DCE | — | 25 | 5 | 80 |
| 17 | 4a | Toluene | — | 25 | 5 | 74 |
| 18 | 4a | Dioxane | — | 25 | 5 | 76 |
| 19 | 5a | H2O | — | 25 | 2 | 71 (76)e |
| 20 | 5a | H2O | InCl3 | 25 | 2 | 68 |

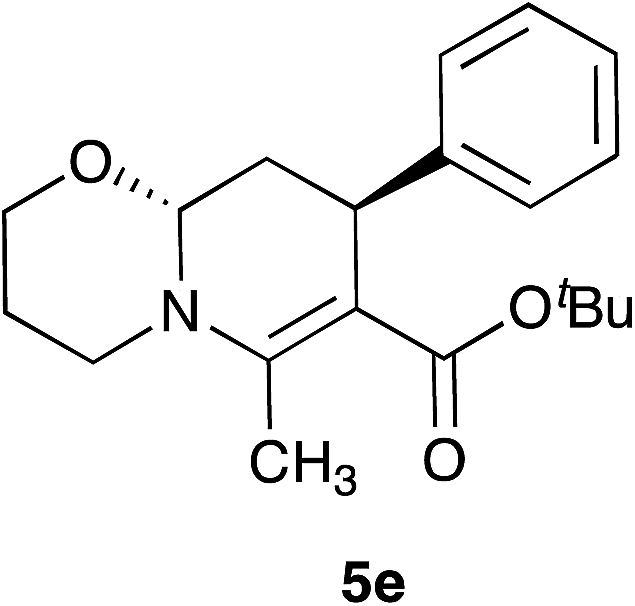
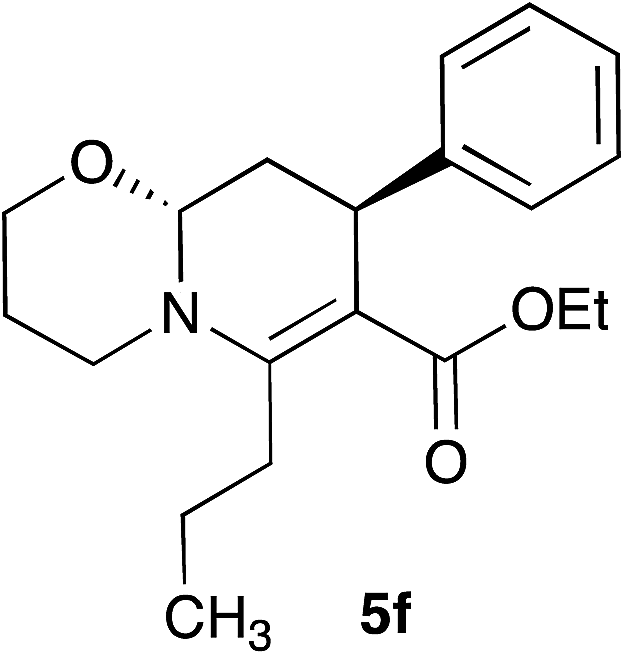
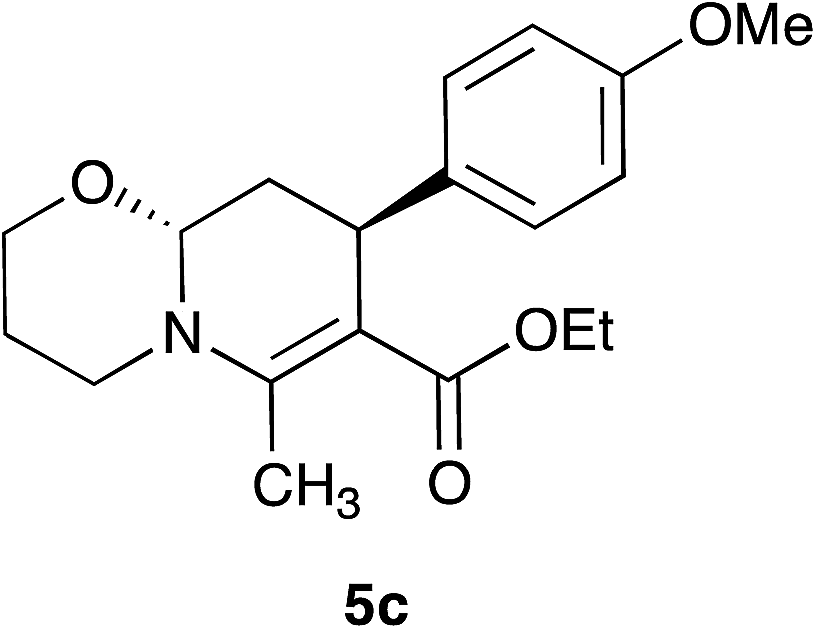
With the optimal reaction conditions in hand, we investigated the scope and limitations of the methodology. At the outset, the substituted cinnamaldehydes 3 bearing both electron-releasing and withdrawing groups were synthesized in good yields involving a Heck reaction between aryl halides and acrolein diethyl acetal in the presence of palladium acetate followed by acid-catalyzed deprotection.26 A number of 1,3-dicarbonyl compounds 2, and α,β-unsaturated aryl aldehydes 3 were employed combining with ethanolamine 1a and 3-propanolamine 1b to access a variety of oxazolo[3,2-a]pyridines 4 and pyrido[2,1-b][1,3]oxazines 5 (Table 2). In most cases the products were obtained as a single trans diastereomer, however, in some cases small amounts (<10%) of the cis diastereomer was observed in the crude 1H-NMR spectra. Although ethyl and t-butyl acetoacetates afforded the products in good yields, ethyl benzoylacetate was found to be less efficient (62–65% yield, entry 7 and 17). Besides β-ketoesters, 1,3-diketones were also effective furnishing the products in good yields (entries 8, 15 and 16). The reaction also tolerated alkyl substituents at C-2 position apart from methyl group (entries 5 and 14). Cinnamaldehyde derivatives bearing both electron-donating (Me, OMe) and withdrawing groups (F) afforded the corresponding products without significant change in their reactivities.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Product (4, 5) | Yieldb (%) | Entry | Product (4, 5) | Yieldb (%) | Entry | Product (4, 5) | Yieldb (%) |
| a Reaction conditions: unless otherwise noted, all reactions were carried out with 1 (1.3 mmol), 2 (1 mmol) and 3 (1 mmol) in 1 mL water for 2 h at 25 °C without any catalyst. b Isolated yield. | ||||||||
| 1 |

|
75 | 7 |
|
62 | 13 |
|
81 |
| 2 |

|
83 | 8 |

|
61 | 14 |
|
70 |
| 3 |

|
80 | 9 |

|
73 | 15 |

|
78 |
| 4 |

|
73 | 10 |

|
77 | 16 |

|
74 |
| 5 |

|
81 | 11 |
|
78 | 17 |

|
65 |
| 6 |

|
82 | 12 |

|
78 | |||

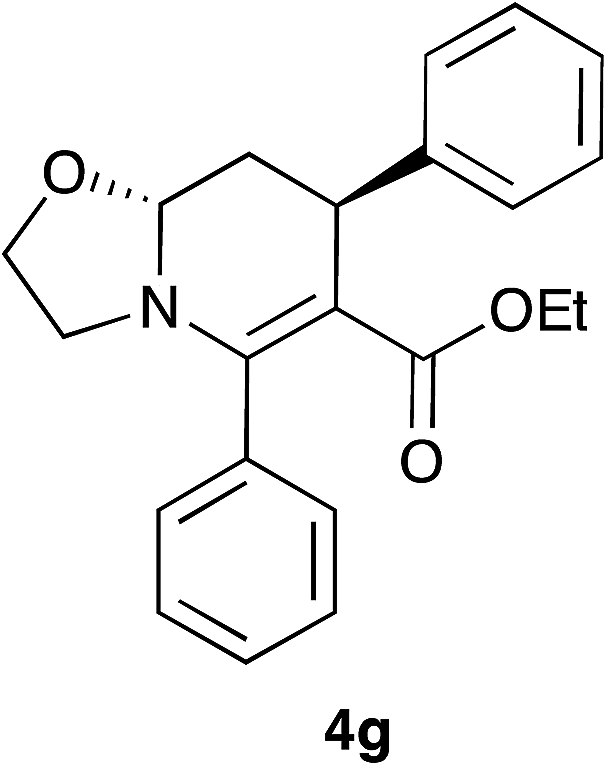
The observed trans diastereoselectivity of compounds 4 and 5 were unambiguously assigned with the help of 1H-NMR coupling constant values of a representative compound 4g. The Ha, Hb, Hc and Hd hydrogens of compound 4g appeared at 4.55 (dd, J = 10.2, 3.6 Hz), 2.28 (ddd, J = 12.3, 3.6, 2.4 Hz), 1.74 (ddd, J = 12.0, 10.2, 5.7 Hz) and 4.32 (dd, J = 5.7, 2.4 Hz) ppm respectively (Fig. 1). The coupling constants of Ha confirmed its axial position owing to the presence of a diaxial coupling with hydrogen Hc (10.2 Hz) besides the axial-equatorial coupling with Hb (3.6 Hz). On the other hand, Hd occupied the equatorial position since no large diaxial coupling was observed with hydrogens Hb and Hc (J = 5.7 and 2.4 Hz). If this hydrogen was present in the axial position (cis isomer), a relatively large diaxial coupling would have observed with the axial hydrogen Hc. Consequently, it is confirmed that the Ha and Hd hydrogens are trans to each other.
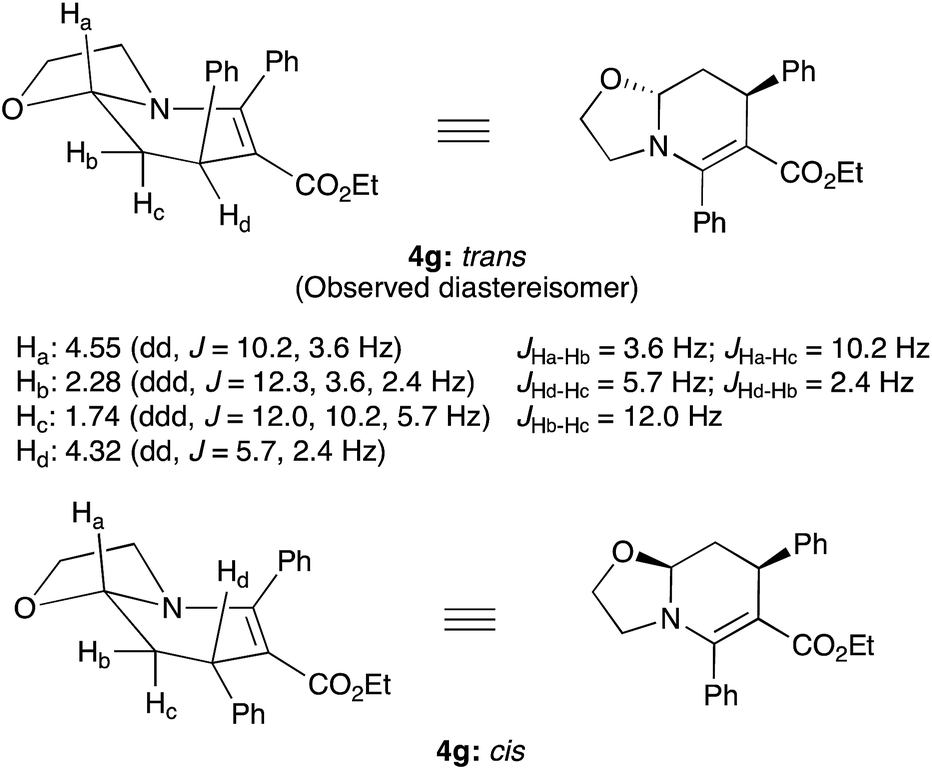 | ||
| Fig. 1 Trans diastereoselectivity assignment based on 1H-NMR coupling constants. | ||
Finally, to further explore the scope of this methodology, we investigated the use of cyclic 1,3-diketones to obtain [1,3]oxazino[3,2-a]quinolin-7(1H)-one 7 and oxazolo[3,2-a]quinolin-6(2H)-one 8 (Scheme 1). Unexpectedly, treatment dimedone 6 with 3-propanolamine 1b and cinnamaldehyde 3a under optimized conditions (water, 25 °C, 3 h) afforded only traces of the product 7 together with 22% of the side product 9. Increase of the reaction temperature to 80 °C did not improve the reaction significantly, and furnished merely 12% of the product. Subsequently we found that the use of 10 mol% of CAN as a catalyst in acetonitrile triggered the reaction to yield 42% of the product again with 21% of compound 9. Under similar conditions 27% of oxazolo[3,2-a]quinolin-6(2H)-one 8 was isolated using ethanolamine as the starting material. Modification of reaction conditions including temperature (80 °C), solvent (DCM and EtOH) and catalyst (InCl3) was not effective to improve the yields of 7 and 8 considerably.
 | ||
| Scheme 1 Synthesis of [1,3]oxazino[3,2-a]quinolin-7(1H)-one 7 and oxazolo[3,2-a]quinolin-6(2H)-one 8. | ||
We also investigated the possibility of utilizing o-aminophenols to access the benz-fused oxazolo[3,2-a]pyridines 12. However, in the reaction between o-aminophenols 10, ethyl acetoacetate 2a and cinnamaldehyde 3a under our optimized conditions the sole isolated product was imine 11 derived from compounds 10 and 3a (Scheme 2). Modifications of reactions conditions including temperature, solvent and use of Lewis acid catalyst were unsuccessful to obtain the expected product.
 | ||
| Scheme 2 Attempts to benz-fused oxazolo[3,2-a]pyridines. | ||
We have proposed a mechanism for the three-component reaction between amino alcohols 1, 1,3-dicarbonyl compounds 2 and α,β-unsaturated aryl aldehydes 3 involving sequential enamine formation, Michael additions, intramolecular cyclization and intramolecular iminium ion cyclization steps. Initial reaction between amino alcohols 1 and 1,3-dicarbonyl compounds 2 affords the enamine intermediate A, which undergoes Michael addition with compounds 3 to furnish intermediate B generating the initial C–N and C–C bonds.21,22 Subsequent intramolecular cyclization followed by dehydration affords iminium ion D through the intermediacy of 6-hydroxy-1,4,5,6-tetrahydropyridine C with the second new C–N bond. Final intramolecular nucleophilic cyclization of species D provides oxazolo[3,2-a]pyridines 4 and pyrido[2,1-b][1,3]oxazines 5. The observed trans stereochemistry could be explained through the attack of the hydroxyl nucleophile in the iminium ion D′ from the opposite side of the aryl substituent.22b,19 Formation of product 5a in 76% yield starting from isolated enamine A, derived from 3-propanolamine 1b and ethyl acetoacetate 2a, and cinnamaldehyde 3a under the optimized reaction conditions supports the enamine formation-initiated mechanism (Scheme 3).
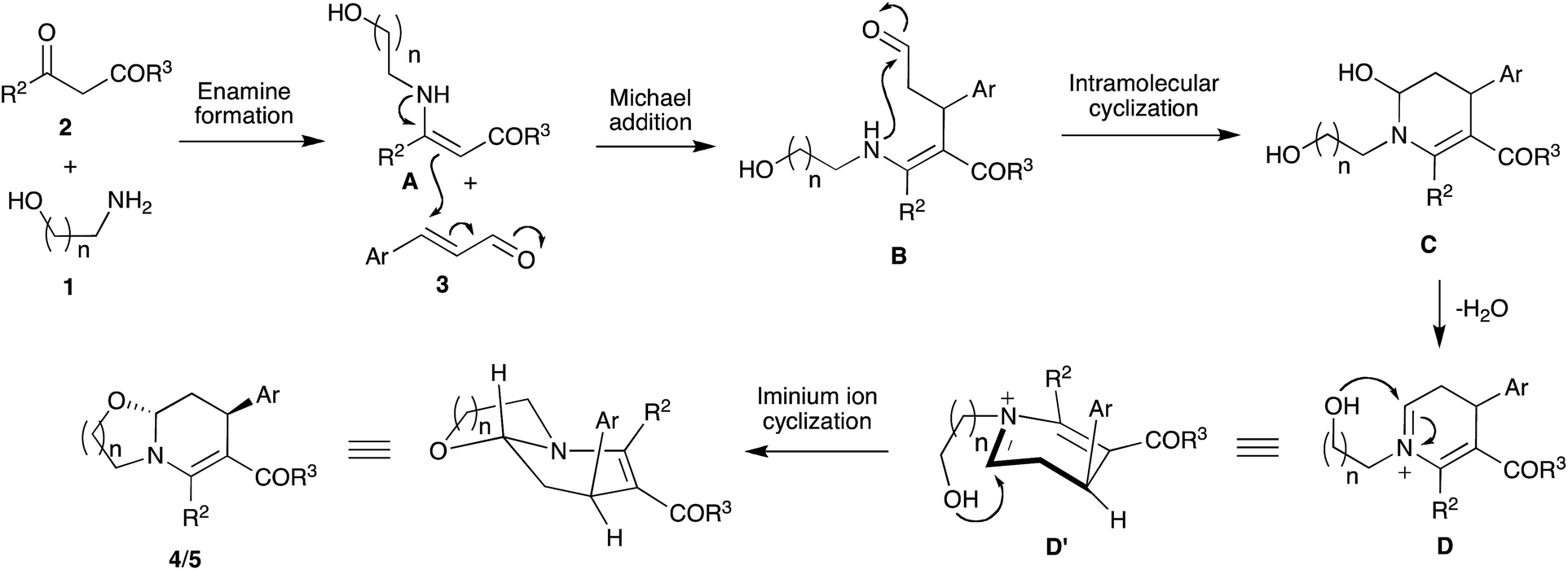 | ||
| Scheme 3 Proposed enamine-formation initiated mechanism. | ||
Conclusions
In conclusion, we have developed an environmentally benign, three-component protocol for the synthesis of oxazolo[3,2-a]pyridines and pyrido[2,1-b][1,3]oxazines starting from readily available starting materials. The reaction between amino alcohols, 1,3-dicarbonyl compounds and α,β-unsaturated aryl aldehydes in water and in the absence of any catalyst afforded the products in good yields. The reaction was found to be highly diastereoselective affording the trans products exclusively in most of the cases. This procedure is also highly atom- and step-economical since only two molecules of water were obtained as the side product, and two heterocyclic rings were constructed by creating four new bonds (one C–C, two C–N and one C–O) in a single operation. The reaction proceeded via a domino enamine formation, Michael addition, intramolecular cyclization and iminium ion cyclizations sequence.Experimental
General
All reagents and solvents were purchased from commercial suppliers (Avra, Alfa Aesar, Sigma-Aldrich, CDH) and used without further purification. The reactions were monitored by thin-layer chromatography using Merck silica gel 60 F254 and visualized by UV detection or using p-anisaldehyde stain or molecular iodine. Melting points were recorded on a melting point apparatus in capillaries and are uncorrected. 1H- and 13C-NMR spectra were recorded in CDCl3 or DMSO-d6 at room temperature on a Bruker Avance 300 spectrometer operating at 300 MHz for 1H and 75 MHz for 13C. Chemical shifts (δ) are expressed in ppm using TMS as internal standard and coupling constants (J) are given in Hz. Infrared (IR) spectra were obtained in an Agilent Cary630 FTIR spectrometer with a diamond ATR accessory for solid and liquid samples, requiring no sample preparation and the major frequencies were reported in cm−1. Elemental analyses were determined at the CAI de Microanálisis Elemental, Universidad Complutense, by using a Leco 932 CHNS combustion microanalyzer.General procedure for the synthesis of oxazolo[3,2-a]pyridines 4 and pyrido[2,1-b][1,3]oxazines 5
To a stirred suspension of amino alcohol 1 (1.3 mmol) in water (1 mL) was added 1,3-dicarbonyl compound 2 (1.0 mmol) followed by α,β-unsaturated arylaldehyde 3 (1.0 mmol). The resulting mixture was allowed to stir at 25 °C for 2 h. After completion of the reaction, as indicated by TLC, the reaction mixture was diluted with dichloromethane and then washed with water followed by brine. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica column chromatography using pet ether–ethyl acetate mixture as eluent (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v).
:10 v/v).
Characterization data for representative compounds
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ), 1.64–1.73 (m, 1H, Hc), 2.27–2.30 (dt, J = 12.6, 3.3 Hz, 1H, Hb), 2.59 (s, 3H, CH3), 3.52–3.63 (m, 2H, NCH2), 3.82–3.88 (m, 1H, O
), 1.64–1.73 (m, 1H, Hc), 2.27–2.30 (dt, J = 12.6, 3.3 Hz, 1H, Hb), 2.59 (s, 3H, CH3), 3.52–3.63 (m, 2H, NCH2), 3.82–3.88 (m, 1H, O![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) CH2N), 3.94 (q, J = 7.2 Hz, 2H, OCH3), 4.17–4.22 (m, 2H, OCH2N & Ph), 4.39 (dd, J = 10.5, 3.6 Hz, 1H, Hd), 7.11–7.17 (m, 3H, ArH), 7.22–7.26 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3): δ 14.3 (CH3), 18.0 (CH3), 33.5 (PhH), 38.1 (PhCHH2), 46.3 (NH2), 58.7 (COOH2), 65.6 (OH2), 84.5 (OHN), 95.5 (C
CH2N), 3.94 (q, J = 7.2 Hz, 2H, OCH3), 4.17–4.22 (m, 2H, OCH2N & Ph), 4.39 (dd, J = 10.5, 3.6 Hz, 1H, Hd), 7.11–7.17 (m, 3H, ArH), 7.22–7.26 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3): δ 14.3 (CH3), 18.0 (CH3), 33.5 (PhH), 38.1 (PhCHH2), 46.3 (NH2), 58.7 (COOH2), 65.6 (OH2), 84.5 (OHN), 95.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO2Et), 125.8 (ArH), 127.5 (ArH), 128.1 (ArH), 146.5 (Ar-quaternary), 152.5 (NCCO2Et), 168.7 (O). Anal. calcd for C17H21NO3: C, 71.06; H, 7.37; N, 4.87. Found: C, 70.77; H, 7.28; N, 4.75.
CO2Et), 125.8 (ArH), 127.5 (ArH), 128.1 (ArH), 146.5 (Ar-quaternary), 152.5 (NCCO2Et), 168.7 (O). Anal. calcd for C17H21NO3: C, 71.06; H, 7.37; N, 4.87. Found: C, 70.77; H, 7.28; N, 4.75.
Acknowledgements
Financial support from the Department of Science and Technology, DST (No. SB/FT/CS-006/2013) and the Council of Scientific and Industrial Research, CSIR (No. 02(0219)/14/EMR-II) is gratefully acknowledged. JCM acknowledges financial support from MICINN, grant CTQ2012-33272-BQU.Notes and references
- For selected reviews on multicomponent reactions for the synthesis of heterocyclic compounds, see: (a) U. K. Sharma, N. Sharma, D. D. Vachhani and E. V. van der Eycken, Chem. Soc. Rev., 2015, 44, 1836 RSC; (b) C. Allais, J.-M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829 CrossRef CAS PubMed; (c) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633 RSC; (d) R. P. Gore and A. P. Rajput, Drug Invent. Today, 2013, 5, 148 CrossRef CAS; (e) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402 RSC; (f) B. B. Touré and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef PubMed; (g) E. Ruijter and R. V. A. Orru, Synthesis of heterocycles via multicomponent reactions, Springer Verlag, 2010, vol. 1 and 2 Search PubMed.
- For selected multicomponent synthesis of simple five and six membered nitrogen heterocycles, see: (a) T. Vivekanand, P. Vinoth, B. Agieshkumar, N. Sampath, A. Sudalai, J. C. Menéndez and V. Sridharan, Green Chem., 2015, 17, 3415 RSC; (b) F. Tamaddon and M. Farahi, Synlett, 2013, 1791 CrossRef CAS; (c) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Commun., 2013, 49, 591 RSC; (d) O. A. Attanasi, G. Favi, F. Mantellini, G. Moscatelli and S. Santeusanio, Adv. Synth. Catal., 2011, 353, 1519 CrossRef CAS; (e) K. Wang and A. Dömling, Chem. Biol. Drug Des., 2010, 75, 277 CrossRef CAS PubMed; (f) V. P. A. Raja, G. Tenti, S. Perumal and J. C. Menéndez, Chem. Commun., 2014, 50, 12270 RSC; (g) G. Tenti, M. T. Ramos and J. C. Menéndez, Curr. Org. Synth., 2013, 10, 646 Search PubMed; (h) V. S. Aulakh and M. A. Ciufolini, J. Am. Chem. Soc., 2011, 133, 5900 CrossRef CAS PubMed; (i) Z. Zhou, T. Kijima, S. Kuwahara, M. Watanabe and T. Izumi, Tetrahedron Lett., 2008, 49, 3757 CrossRef; (j) S. Tu, T. Li, F. Shi, F. Fang, S. Zhu, X. Wei and Z. Zong, Chem. Lett., 2005, 34, 732 CrossRef CAS; (k) T. Shintani, H. Kadono, T. Kikuchi, T. Schubert, Y. Shogase and M. Shimazaki, Tetrahedron Lett., 2003, 44, 6567 CrossRef CAS.
- (a) C. D. Duffy, P. Maderna, C. McCarthy, C. Loscher, C. Godson and P. J. Guiry, ChemMedChem, 2010, 5, 517 CrossRef CAS PubMed; (b) R. B. Lacerda, C. K. F. de Lima, L. L. da Silva, N. C. Romeiro, A. L. P. Miranda, E. J. Barreiro and C. A. M. Fraga, Bioorg. Med. Chem., 2009, 17, 74 CrossRef CAS PubMed.
- B. Vacher, B. Bonnaud, P. Funes, N. Jubault, W. Koek, M.-B. Assié, C. Cosi and M. Kleven, J. Med. Chem., 1999, 42, 1648 CrossRef CAS PubMed.
- M. Horiuch, C. Murakami, N. Fukamiya, D. Yu, T.-H. Chen, K. F. Bastow, D.-C. Zhang, Y. Takaishi, Y. Imakura and K.-H. Lee, J. Nat. Prod., 2006, 69, 1271 CrossRef CAS PubMed.
- K. K. Borowicz, M. Gasior, Z. Kleinrok and S. J. Czuczwar, Eur. J. Pharmacol., 1997, 323, 45 CrossRef CAS PubMed.
- G. M. Buckley, N. Cooper, R. J. Davenport, H. J. Dyke, F. P. Galleway, L. Gowers, A. F. Haughan, H. J. Kendall, C. Lowe, J. G. Montana, J. Oxford, J. C. Peake, C. L. Picken, M. D. Richard, V. Sabin, A. Sharpe and J. B. H. Warneck, Bioorg. Med. Chem. Lett., 2002, 12, 509 CrossRef CAS PubMed.
- N. Edraki, A. R. Mehdipour, M. Khoshneviszadeh and R. Miri, Drug Discovery Today, 2009, 14, 1058 CrossRef CAS PubMed.
- For reviews on synthesis of pyridine and dihydropyridines, see: (a) M. D. Hill, Chem.–Eur. J., 2010, 16, 12052 CrossRef CAS PubMed; (b) J.-P. Wan and Y. Liu, RSC Adv., 2012, 2, 9763 RSC; (c) Ref. 1b .
- For the biological importance of 1,2,3,4-tetrahydropyridines, see: (a) L. Tesoriere, M. Fazzari, F. Angileri, C. Gentile and M. A. Livrea, J. Agric. Food Chem., 2008, 56, 10487 CrossRef CAS PubMed; (b) S. Nara, R. Tanaka, J. Eishima, M. Hara, Y. Takahashi, S. Otaki, R. J. Foglesong, P. F. Hughes, S. Turkington and Y. Kanda, J. Med. Chem., 2003, 46, 2467 CrossRef CAS PubMed.
- For the synthesis of 1,2,3,4-tetrahydropyridines, see: (a) S. Sun, C. Cheng, J. Yang, A. Taheri, D. Jiang, B. Zhang and Y. Gu, Org. Lett., 2014, 16, 4520 CrossRef CAS PubMed; (b) N. Gigant, G. Dequirez, P. Retailleau, I. Gillaizeau and P. Dauban, Chem.–Eur. J., 2012, 18, 90 CrossRef CAS PubMed; (c) S. Tong, X. Yang, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, Tetrahedron, 2012, 68, 6492 CrossRef CAS.
- A. San Feliciano, E. Caballero, P. Puebla, J. A. P. Pereira, J. Gras and C. Valenti, Eur. J. Med. Chem., 1992, 27, 527 CrossRef CAS.
- E. Caballero, J. I. Manzano, P. Puebla, S. Castanys, F. Gamarro and A. San Feliciano, Bioorg. Med. Chem. Lett., 2012, 22, 6272 CrossRef CAS PubMed.
- (a) E. Martin, M. A. Sevilla, A. Moran, M. L. Martin and L. San Roman, J. Auton. Pharmacol., 2001, 21, 85 CrossRef CAS PubMed; (b) E. Martín, A. Morán, M. L. Martín, L. San Román, P. Puebla, M. Medarde, E. Caballero and A. San Feliciano, Bioorg. Med. Chem. Lett., 2000, 10, 319 CrossRef; (c) A. Moran, E. Martin, C. Velasco, M. L. Martin, L. San Roman, E. Caballero, P. Puebla, M. Medarde and A. San Feliciano, J. Pharm. Pharmacol., 1997, 49, 421 CrossRef CAS PubMed.
- (a) M. Amat, E. Brunaccini, B. Checa, M. Perez, N. Llor and J. Bosch, Org. Lett., 2009, 11, 4370 CrossRef CAS PubMed; (b) M. Amat, C. Escolano, O. Lozano, A. Gómez-Esqué, R. Griera, E. Molins and J. Bosch, J. Org. Chem., 2006, 71, 3804–3815 CrossRef CAS PubMed; (c) C. Escolano, M. Amat and J. Bosch, Chem.–Eur. J., 2006, 12, 8198 CrossRef CAS PubMed.
- For pyrido[2,1-b][1,3]oxazines related compounds, see: (a) J. Sun, H. Gong and C.-G. Yan, Tetrahedron, 2013, 69, 10235 CrossRef CAS; (b) S. Asghari and A. K. Habibi, Tetrahedron, 2012, 68, 8890 CrossRef CAS; (c) A. A. Esmaeili, H. Vesalipoor, R. Hosseinabadi, A. F. Zavareh, M. A. Naseri and E. Ghiamati, Tetrahedron Lett., 2011, 52, 4865 CrossRef CAS; (d) I. Hermecz, Adv. Heterocycl. Chem., 2003, 85, 173 CrossRef CAS.
- For oxazolo[3,2-a]pyridine related compounds, see: (a) V. B. Rybakov and E. V. Babaev, Chem. Heterocycl. Compd., 2014, 50, 225 CrossRef CAS; (b) I. V. Ukrainets, E. V. Mospanova, N. A. Jaradat, O. V. Bevz and A. V. Turov, Chem. Heterocycl. Compd., 2012, 48, 1347 CrossRef CAS; (c) I. V. Ukrainets, E. V. Mospanova, O. V. Gorokhova and S. V. Shishkina, Chem. Heterocycl. Compd., 2011, 47, 1014 CrossRef CAS; (d) D. Gnecco, A. M. Lumbreras, J. L. Teran, A. Galindo, J. R. Juarez, M. L. Orea, A. Castro, R. G. Enriquez and W. F. Reynolds, Heterocycles, 2009, 78, 2589 CrossRef CAS; (e) E. V. Babaev, V. L. Alifanov and A. V. Efimov, Russ. Chem. Bull., 2008, 57, 845 CrossRef CAS; (f) A. Castro, J. Ramirez, J. Juarez, J. L. Teran, L. Orea, A. Galindo and D. Gnecco, Heterocycles, 2007, 71, 2699 CrossRef CAS; (g) A. Bush and E. V. Babaev, Molecules, 2003, 8, 460 CrossRef CAS.
- (a) E. Caballero, P. Puebla, M. Medarde, M. Sánchez, M. A. Salvadó, S. García-Granda and A. San Feliciano, J. Org. Chem., 1996, 61, 1890 CrossRef CAS PubMed; (b) E. Caballero, P. Puebla, M. Medarde and A. San Feliciano, Tetrahedron, 1993, 49, 10079 CrossRef CAS; (c) A. San Feliciano, E. Caballero, J. A. P. Pereira and P. Puebla, Tetrahedron, 1991, 47, 6503 CrossRef CAS.
- J.-P. Wan, Y. Lin, Q. Huang and Y. Liu, J. Org. Chem., 2014, 79, 7232 CrossRef CAS PubMed.
- R. Nöel, C. Vanucci-Bacqué, M.-C. Fargeau-Bellassoued and G. Lhommet, J. Org. Chem., 2005, 70, 9044 CrossRef PubMed.
- (a) V. Sridharan, S. Maiti and J. C. Menéndez, Chem.–Eur. J., 2009, 15, 4565 CrossRef CAS PubMed; (b) V. Sridharan, S. Maiti and J. C. Menéndez, J. Org. Chem., 2009, 74, 9365 CrossRef CAS PubMed.
- For representative examples, see: (a) P. A. Suryavanshi, V. Sridharan, S. Maiti and J. C. Menéndez, Chem.–Eur. J., 2014, 20, 8791 CrossRef CAS PubMed; (b) P. A. Suryavanshi, V. Sridharan and J. C. Menéndez, Chem.–Eur. J., 2013, 19, 13207 CrossRef CAS PubMed; (c) S. Maiti, V. Sridharan and J. C. Menéndez, J. Comb. Chem., 2010, 12, 713 CrossRef CAS PubMed.
- R. Nöel, M.-C. Fargeau-Bellassoued, C. Vanucci-Bacqué and G. Lhommet, Synthesis, 2008, 1948 Search PubMed.
- For a review on catalyst-free in-water, on-water organic synthesis, see: M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- For a review on CAN-catalyzed organic synthesis, see: V. Sridharan and J. C. Menéndez, Chem. Rev., 2010, 110, 3805 CrossRef CAS PubMed.
- G. Battistuzzi, S. Cacchi and G. Fabrizi, Org. Lett., 2003, 5, 777 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and copies of spectra of products. See DOI: 10.1039/c5ra18804k |
| This journal is © The Royal Society of Chemistry 2015 |