
Imidazolium-supported benzotriazole: an efficient and recoverable activating reagent for amide, ester and thioester bond formation in water†‡
Abstract
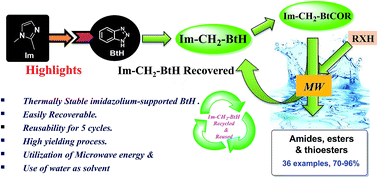
An efficient and recyclable imidazolium-supported benzotriazole reagent (Im-CH2-BtH) as a novel synthetic auxiliary has been synthesized and its utility as a carboxyl group activating reagent via the formation of stable imidazolium-supported acyl benzotriazoles was explored for the synthesis of amides, esters and thioesters in water under microwave conditions. The reagent was reused five times without any noticeable loss in activity. It is moisture insensitive and highly stable under thermal and aerobic conditions. The application of imidazolium-supported N-acetyl benzotriazole leads to synthesis of paracetamol on the gram scale under greener conditions in 93% yield.
 Please wait while we load your content...
Please wait while we load your content...

