
Preparation and characterization of pullulanase debranched starches and their properties for drug controlled-release
Abstract
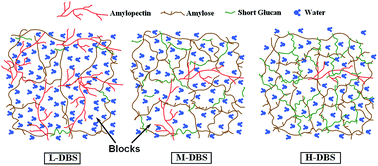
Debranched starches (DBSs) with different degrees of debranching (low, L-DBS; moderate, M-DBS; high, H-DBS) were prepared and investigated. After pullulanase modification, the starch granules became more porous and many small particles containing short glucan chains were generated. DBSs adopt a single-helical V-type crystalline structure with low crystallinity. L-DBS samples contained fewer (20.70%) and longer (degree of polymerization, DP: 21.92) linear short glucan chains than their counterparts (M-DBS: 40.92%, 20.05 DP; H-DBS: 55.52%, 18.52 DP). Pullulanase enzymatic hydrolyzate for DBS samples with higher degrees of debranching inclined towards retrogradation at 20 °C. DBSs with higher degrees of debranching could form a hydrogel with higher G′ and G′′ values, indicating these samples formed a stronger gel network. L-DBS could hold more water and its digestibility was higher. The in vitro test showed that DBS is a good candidate to control drug release for over 12 h. Furthermore, the drug release profiles from both DBS-based and HPMC-based tablets showed an anomalous transport mechanism. The drug release from these four matrices was controlled by a combination of drug diffusion and matrix erosion. The drug release properties from DBS-based tablets were considerably influenced by the degree of debranching. The in vitro drug release profile of M-DBS was similar to that of HPMC (f2 = 60.75), while L-DBS and H-DBS differed from HPMC (f2 < 50). In summary, DBS is a good hydrogel candidate, and it can be used as an excipient in oral tablets to control drug release.
 Please wait while we load your content...
Please wait while we load your content...

