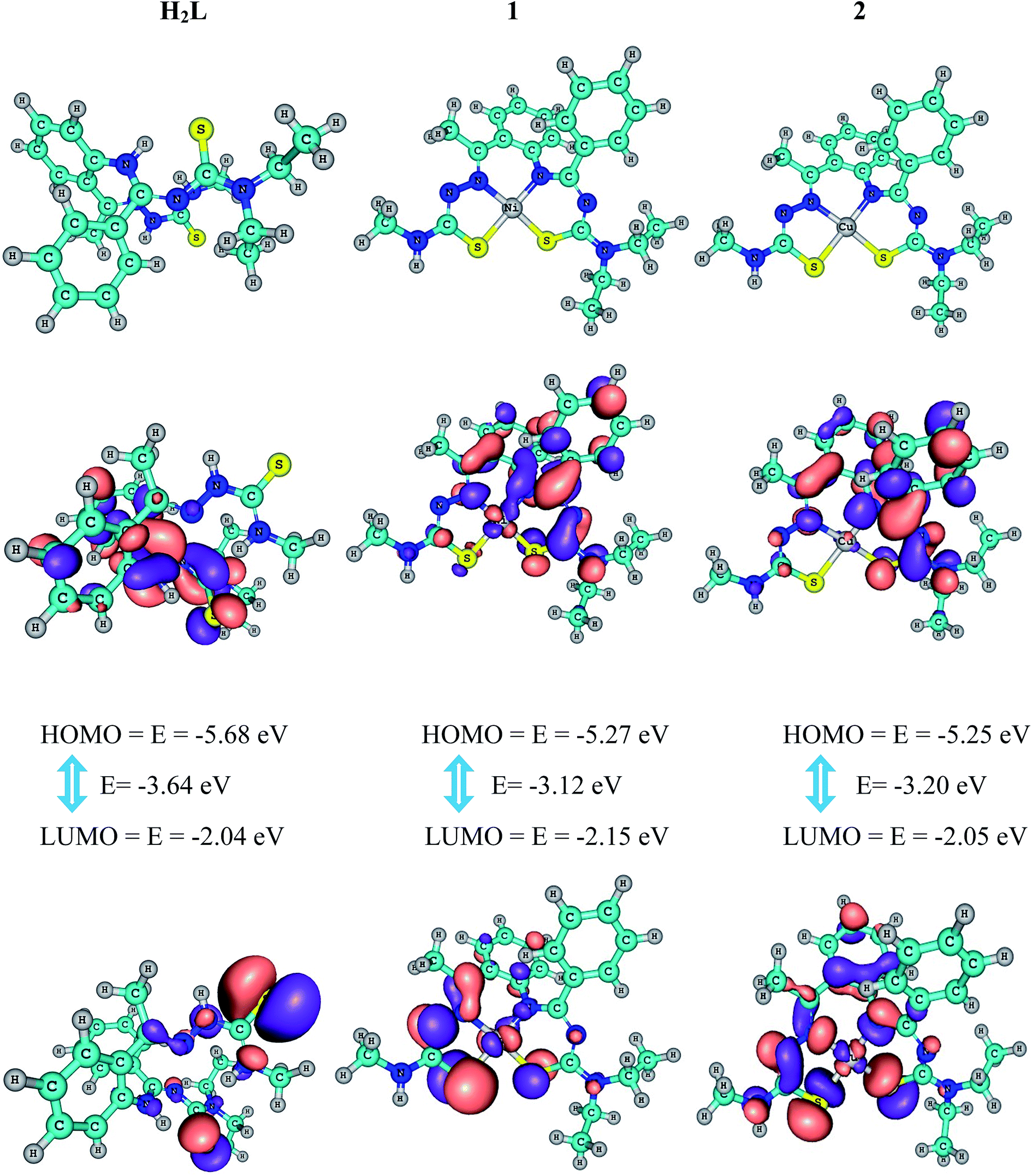DOI:
10.1039/C5RA18568H
(Paper)
RSC Adv., 2015,
5, 103321-103342
Nickel(II) and copper(II) complexes constructed with N2S2 hybrid benzamidine–thiosemicarbazone ligand: synthesis, X-ray crystal structure, DFT, kinetico-catalytic and in vitro biological applications†
Received
10th September 2015
, Accepted 17th November 2015
First published on 18th November 2015
Abstract
With the aim of assessing whether transition metal complexes might be utilized as efficient biocatalysts and biological drugs, new monofunctional nickel(II) and copper(II) complexes of types NiL (1) and CuL (2) [H2L = N-(N′,N′-diethylaminothiocarbonyl)benzimidoyl chloride-2-aminoacetophenone-N-methylthiosemicarbazone] were synthesized. The compounds are remarkably stable and were obtained in good yields. Structural elucidation was carried out thoroughly in solid and solution-states by elemental analysis and various spectroscopy techniques (IR, UV-vis, 1H NMR, and EPR) as well as ESI mass spectrometry. The molecular structure of the compounds was investigated by single-crystal X-ray diffraction. The analyses showed tetradentate coordination of the ligand in nickel(II) and copper(II) complexes, in which the metal atom exhibits a square planar geometry with N2S2 in a donor fashion. Structural optimization, HOMO–LUMO energy calculations and Natural Bond Orbital (NBO) analysis of H2L and its complexes (1 and 2) were investigated by Density Functional Theory (DFT). The trend in binding affinities of the compounds with biomolecules, such as calf thymus DNA (CT-DNA) and bovine serum albumin (BSA) protein, were investigated by different spectrophotometric methods, which revealed an intercalative mode of interaction. Furthermore, enzyme kinetic studies reflected that the square planar complexes (1 and 2) are also effective in mimicking catecholase (3,5-DTBC) and phosphatase (4-NPP) activities over the parent H2L. The high kcat values suggested that the selected compounds displayed a high rate of catalytic efficiency. In vitro cytotoxicity of the complexes on human skin cancer melanoma (A375), human cervical cancer (HeLa) and human hepatocellular carcinoma (Hep3B) cancer cell lines demonstrated that the complexes had a broad-spectrum of anti-cancer activity with low IC50 values. The morphological assessment data obtained by acridine orange/ethidium bromide (AO/EB) and diamidino-2-phenylindole (DAPI) staining revealed that complex 2 induces apoptosis much more effectively than 1.
Introduction
Ligand design is becoming an increasingly important part of synthetic activity in chemistry. This is because of the subtle control that ligands exert on the metal center to which they are coordinated.1,2 Notably, the choice of hybrid ligands is particularly crucial in the stabilization of highly reactive species, promoting reactivity in unusual oxidation states3,4 and implementing catalytic as well as biological properties.5,6 Previously, many articles have shown that mono and bidentate ligand systems may have insufficient stability for in vitro and in vivo studies, due to rapid ligand exchange reactions with plasma components. However, many metal(II) cores containing medium and soft donor atom ligands were particularly recommended for in vitro and in vivo studies. Thus, chelators with a mixed sulfur and nitrogen donor sphere might be very suitable for the formation of metal complexes and some of them are used in medicinal applications.7 With the abovementioned fact in mind, tetradentate N, S donor Schiff-base ligands have extensively been used as supporting ligands in d-block chemistry because of their ability to stabilize metals in different oxidation states.8,9
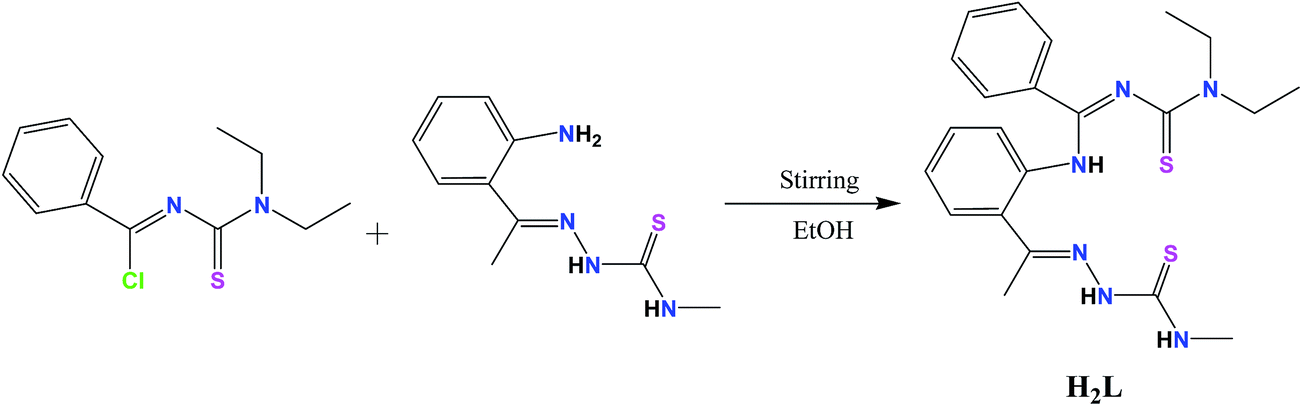
Thioureas are potentially very versatile ligands, as they are able to coordinate to a range of metal centers as neutral ligands, monoanions, or dianions. The effectiveness of thiourea is believed to be due to the presence of a lone pair of electrons in atoms, such as nitrogen, sulfur and oxygen and π-electron clouds, which are capable of forming dative covalent bonds with metals.10 Derivatives of thiourea such as N-(dialkylaminothiocarbonyl)benzimidoyl chlorides were first synthesized and incorporated into the synthesis of the corresponding benzamidines, which can readily be obtained by reactions of benzimidoyl chlorides and primary amines, with numerous metals that have been broadly studied.11 Benzamidines can be functionalized with an additional donor group to modify the properties, depending on the requirements. Thiosemicarbazones have been chosen to functionalize benzimidoyl chlorides due to their versatile coordination behavior and promising biological and pharmaceutical properties.12–15 A new tetradentate chelator has been synthesized from a benzimidoyl chloride moiety and N-methylthiosemicarbazide (Scheme 1).
 |
| | Scheme 1 Synthetic route of H2L in this study. | |
The discovery of cisplatin in 1965 was a major milestone in cancer chemotherapy; cisplatin forms a Pt-DNA adduct and ultimately inhibit cellular DNA transcription and replication. However, cisplatin causes side effects such as drug resistance, and severe toxicity, for example, nephrotoxicity and neurotoxicity.16–18 Therefore, new metal-based drugs with novel mechanisms of action are being developed as an alternative to platinum-based drugs. Numerous Co(III), Ni(II), Cu(I/II), Zn(II) and Ru(II) complexes have been probed in the quest for new inorganic drugs.19a–c Among the various metal(II) complexes, nickel and copper ions are considered as the most promising alternatives to cisplatin as anticancer drugs because they play an energetic role in biological functions such as DNA, BSA, HSA and RNA interactions.19d
To find highly effective, target-specific and less toxic drugs, much effort has been devoted for the development of metal-based anticancer agents that bind to DNA through a noncovalent interaction.20 There are three noncovalent binding modes of metal complexes with DNA: electrostatic, groove, and intercalation. Many important applications of these complexes require that they can bind to DNA via an intercalative mode that could induce cellular degradation. Planarity of the ligand and the presence of aromatic systems are factors that favor intercalation.21a Moreover, most of the recent research on clinical agents has shown that drugs (metal complexes) not only bind to the prime molecular target, DNA, but also strongly interact with serum albumin proteins such as BSA and HSA.21b It is essential to explore drug–protein interactions as most of the drugs bound to serum albumin are transported as a protein complex. Attention has also been focused on the proteins that drive and control cell cycle progression.22
On the other hand, the catecholase/phosphatase-like activities of some model coordination complexes have been a topic of recent interest with respect to the development of new bioinspired catalysts.23 Catechol oxidase is a copper-containing type-III active-site protein (Scheme 2) that catalyzes two-electron-transfer reactions during the oxidation of a wide range of catechols, which is known as catecholase activity, whereas hydrolase belongs to a class of metalloenzymes that catalyze the hydrolysis of several substrates, including phosphate esters (4-NPP), peptides and nucleic acids.24a To date, most researchers have focused on dinuclear systems that structurally match the original enzyme, wherein there exists a dimeric-copper active center. Recent investigations have also shown that some manganese(II/III), nickel(II), zinc(II) and cobalt(II/III) species can also mediate such catechol oxidation.24b,c Therefore, much attention has been focused on the catecholase/phosphatase-like properties of benzamidine–thiosemicarbazone-containing nickel(II) and copper(II) complexes.
 |
| | Scheme 2 Bio-active dimeric copper-containing type-III protein. | |
Therefore, through the abovementioned contribution, we reported the use of the ligand [N-(diethylaminothiocarbonyl)]benzimidoylchloride-2-aminoacetophenone-N-methylthiosemicarbazone] (H2L) in the synthesis of new nickel(II) and copper(II) complexes (1 and 2) as shown in Scheme 3. Furthermore, investigation of the binding properties of these complexes with calf thymus DNA (CT-DNA) and BSA protein has been carried out. Moreover, H2L and its complexes, 1 and 2, were also investigated for possible catecholase- and phosphatase-like activities. In addition, the cytotoxic activity of the new compounds towards the normal Vero cell line, A375 (human melanoma skin cancer), human cervical cancer (HeLa) and human hepatocellular carcinoma (Hep3B) cell lines was evaluated using MTT assay along with cell death analysis by AO/EB and DAPI staining.
 |
| | Scheme 3 Synthesis of nickel(II) (1) and copper(II) (2) complexes. | |
Results and discussion
Synthesis and characterization
The benzamidine–thiosemicarbazone ligand, H2L, was synthesized by the reaction of N-(diethylaminothiocarbonyl)benzimidoylchloride10 with o-aminoacetophenone–thiosemicarbazone25 in the presence of the supporting base, Et3N. The syntheses of nickel(II) and copper(II) complexes (1 and 2) were achieved in good yields by the reaction of H2L with Ni(OAc)2·4H2O and Cu(OAc)2·H2O, respectively (Scheme 3). In this reaction, H2L acted as a dibasic tetradentate ligand in N2S2 fashion. The new compounds under investigation are crystalline and nonhygroscopic solids, which are air-stable in solution and in the solid state at room temperature, soluble in common organic solvents, such as methanol, ethanol, benzene, chloroform, dichloromethane, acetone, dimethylsulfoxide, and dimethylformamide, and insoluble in hexane, petroleum ether and diethyl ether. The complexes have been characterized satisfactorily by elemental analysis and IR, UV-vis, 1H-NMR and ESI-MS spectral studies. In addition, the molecular structure of the ligand H2L and its complexes, 1 and 2, was confirmed by single-crystal X-ray crystallography.
IR spectroscopy has been used to confirm the deprotonation and exact binding mode of the ligand. In the IR spectrum of the ligand, three νNH bands were observed in the range of 3236–3302 cm−1.26 On complexation, the two νNH peaks disappeared, indicating deprotonation of these groups in the ligand, and a sharp band at 3390–3417 cm−1 was assigned to the νNH stretch of the H3C–NH–C–S group. A strong vibration was observed at 1668–1717 cm−1 in the spectrum of the ligand, corresponding to νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, which was shifted to lower frequency in the complexes. A sharp band at 838 cm−1 was attributed to νCS of the diethylaminothiocarbonyl moiety in H2L. This was shifted to the lower frequency region 784–790 cm−1 in the complexes, indicating participation of sulfur in the coordination. Furthermore, one more band at 781 cm−1 was assigned to νCS, present in the thiosemicarbazone part of H2L; this completely disappeared in the spectra of the complexes and a new band at around 761–764 cm−1 was assigned to νC–S, indicating thione–thiol tautomerism of the –NH–CS group and subsequent coordination of thiolate sulfur after deprotonation. These results clearly confirm that coordination occurs through nitrogen and sulfur atoms.
N, which was shifted to lower frequency in the complexes. A sharp band at 838 cm−1 was attributed to νCS of the diethylaminothiocarbonyl moiety in H2L. This was shifted to the lower frequency region 784–790 cm−1 in the complexes, indicating participation of sulfur in the coordination. Furthermore, one more band at 781 cm−1 was assigned to νCS, present in the thiosemicarbazone part of H2L; this completely disappeared in the spectra of the complexes and a new band at around 761–764 cm−1 was assigned to νC–S, indicating thione–thiol tautomerism of the –NH–CS group and subsequent coordination of thiolate sulfur after deprotonation. These results clearly confirm that coordination occurs through nitrogen and sulfur atoms.
The clearest characterization of the synthesized compounds can be observed from the 1H-NMR spectra of ligand H2L and its complex 1. The data are summarized in the experimental section and the resulting spectra are depicted in Fig. S1 and S2.† In the 1H NMR spectra, the signal due to the –NH hydrazinic group of the ligand appeared at 12.72 ppm; this was absent in the complex, supporting the ketoenolization and coordination of the thiolate sulfur.27 The signal due to the Car–NH group, which appeared at 8.47 ppm in the spectrum of H2L, completely disappeared in the spectrum of the complex, indicating deprotonation upon complex formation. A signal at 8.10 ppm was attributed to the NH–CH3 group of H2L; this was shifted to high field at 4.85 ppm after complexation.28 However, the proton signals of the two methylene groups in H2L and its complex 1, which should consequently be two quartets, appear as four well-separated multiplet resonances at 3.74–4.12 ppm.6 This results in magnetic nonequivalence with respect to the axial and equatorial positions of the four methylene protons, as indicated by four separated signals with ABX3 splitting patterns as previously reported.29,30 Furthermore, two triplet signals at 1.22–1.33 ppm for the methyl groups in N(CH2CH3)2 are observed in the spectra of H2L and its complex 1.7 In addition, the spectra of H2L and its complex 1 showed a series of overlapping multiplets for aromatic protons at 6.40–7.54 ppm.
UV-vis spectra of the complexes obtained in DMSO solution (0–48 h) (Fig. S3†) are similar, suggesting that the complexes retain their structure in solution within the timeframe used for the biological experiments. Electronic spectra of the compounds displayed intense absorptions in the visible and ultraviolet regions. The absorptions in the ultraviolet region are assignable to transitions within the ligand orbitals. The bands around 236–298 nm were assigned to ligand-centered (LC) π → π* and n → π* transitions.15a The lowest energy absorption maxima located in the 367–374 nm range may be assigned to an S(pπ) → M(dπ) (M = Ni2+, Cu2+) LMCT transition caused by promotion of the electron from the full HOMO of the ligand of primarily sulfur pπ character to the empty LUMO of metal ion dπ character. Furthermore, the complexes showed broad bands in the 431–491 nm region, which are attributed to d–d transitions of spin-paired d8 (complex 1) and d9 (complex 2) species with a square-planar structure (Fig. S3†). The geometry of the complexes was further confirmed by single-crystal X-ray diffraction analysis.
The EPR spectrum of complex 2 measured at 9.45 GHz (X-band) corresponds to an axial g-tensor showing broad signals (Fig. S4†). The g-parameters, g‖ and g⊥, were determined for complex 2 but no coupling parameters could be extracted. The averaged g-values (gav) were obtained through the relationship between the anisotropic parameters of the g-tensors, from the following expression: gav = (g‖ + 2 × g⊥)/3. Complex 2 exhibits an anisotropic spectrum with g‖ = 2.078, g⊥ = 2.031 and gav = 2.046, which is consistent with the presence of a square planar geometry, as is evident from the crystal structure of 2. These parameters are in good agreement with those of the related square planar Cu2+ systems and are typical of axially symmetrical d9 copper(II) complexes15b (Fig. S4†).
The ESI-MS analyses of H2L and complexes 1 and 2 show the most abundant peaks at m/z, 441.55, 498.42, and 503.52, which were assigned to [M + H]+ ions. The molecular masses obtained are in good agreement with the calculated molecular masses (Fig. S5–S7†).
Single-crystal X-ray diffraction studies
Single-crystal X-ray diffraction studies of the compounds (H2L, 1 and 2) confirm the conclusions drawn from the spectroscopy studies. To gain insight into the coordination chemistry and structural parameters of these compounds, good-quality single crystals were isolated by slow evaporation of a concentrated ethanol/dichloromethane solution and characterized by X-ray diffraction. Details of the data collection, solution and refinement are gathered in the experimental section and in Table 1. ORTEP views of the molecular structures of compounds along with partial atom numbering schemes are shown in Fig. 1–3 and important bond lengths and angles for the compounds are summarized in Table 2.
Table 1 Crystal data and structure refinement for ligand H2L and its complexes 1 and 2
| |
H2L |
1 |
2 |
| Empirical formula |
C22H28N6S2 |
C22H26N6NiS2 |
C22H26CuN6S2 |
| Formula weight |
440.62 |
497.32 |
502.15 |
| Temperature (K) |
100(2) K |
295(2) K |
295(2) |
| Wavelength (Å) |
0.71073 Å |
0.71073 Å |
0.71073 Å |
| Crystal system |
Triclinic |
Orthorhombic |
Orthorhombic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca |
Pbca |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Unit cell dimensions |
| a (Å) |
7.831(6) Å |
13.2007(7) Å |
13.3637(4) Å |
| b (Å) |
10.956(8) Å |
17.5298(9) Å |
17.6802(7) Å |
| c (Å) |
14.357(18) Å |
20.1722(11) Å |
19.9696(6) Å |
| α (°) |
75.594(18)° |
90° |
90° |
| β (°) |
89.60(3)° |
90° |
90° |
| γ (°) |
72.16(2)° |
90° |
90° |
| Volume (Å3) |
1132.47(5) Å3 |
4668.0(4) Å3 |
4718.3(3) Å3 |
| Z |
2 |
8 |
8 |
| Density (calcd) mg m−3 |
1.292 mg m−3 |
1.415 mg m−3 |
1.414 mg m−3 |
| Absorption coefficient |
2.291 mm−1 |
1.032 mm−1 |
1.124 mm−1 |
| F(000) |
468 |
2080 |
2088 |
| Crystal size (mm3) |
0.12 × 0.07 × 0.03 |
0.37 × 0.17 × 0.03 |
0.25 × 0.22 × 0.17 |
| Theta range for data collection |
3.18 to 73.12° |
3.44 to 25.04° |
3.608 to 25.07° |
| Index ranges |
−9 ≤ h ≤ 8, |
−15 ≤ h ≤ 15 |
−15 ≤ h ≤ 11 |
| −13 ≤ k ≤ 12 |
−20 ≤ k ≤ 20 |
−21 ≤ k ≤ 14 |
| −17 ≤ l ≤ 17 |
−19 ≤ l ≤ 24 |
−23 ≤ l ≤ 19 |
| Reflections collected |
12690 |
16086 |
13366 |
| Independent reflections |
4299 [R(int) = 0.0182] |
4122 [R(int) = 0.0666] |
4158 [R(int) = 0.0314] |
| Data/restraints/parameters |
4299/0/275 |
4122/0/284 |
4158/0/284 |
| Goodness-of-fit on F2 |
1.132 |
1.059 |
1.049 |
| Final R indices [I > 2 sigma(I)] |
R1 = 0.0360, wR2 = 0.1007 |
R1 = 0.0440, wR2 = 0.1096 |
R1 = 0.0372, wR2 = 0.0910 |
| R indices (all data) |
R1 = 0.0364, wR2 = 0.1011 |
R1 = 0.0607, wR2 = 0.1180 |
R1 = 0.0511, wR2 = 0.0968 |
Table 2 Selected bond lengths (Å) and bond angles (°) for H2L and its complexes, 1 and 2, with the optimized geometrical values
| H2L |
X-ray |
DFT |
1 |
X-ray |
DFT |
2 |
X-ray |
DFT |
| C(1)–N(1) |
1.339(2) |
1.358 |
N(3)–Ni(1) |
1.872(2) |
1.890 |
N(3)–Cu(1) |
1.932(2) |
1.954 |
| C(1)–S(1) |
1.704(2) |
1.784 |
S(1)–Ni(1) |
2.149(9) |
2.262 |
S(1)–Cu(1) |
2.244(7) |
2.365 |
| C(1)–N(2) |
1.362(2) |
1.371 |
N(4)–Ni(1) |
1.900(2) |
1.911 |
N(4)–Cu(1) |
1.981(2) |
1.997 |
| C(2)–N(2) |
1.309(2) |
1.325 |
S(2)–Ni(1) |
2.157(9) |
2.276 |
S(2)–Cu(1) |
2.230(9) |
2.361 |
| C(2)–N(3) |
1.333(3) |
1.356 |
N(3)–C(6) |
1.353(3) |
1.354 |
N(3)–C(2) |
1.353(3) |
1.367 |
| C(9)–N(3) |
1.418(2) |
1.428 |
N(3)–C(13) |
1.409(3) |
1.419 |
N(3)–C(9) |
1.400(3) |
1.412 |
| C(15)–N(4) |
1.274(2) |
1.301 |
N(4)–C(19) |
1.319(4) |
1.315 |
N(4)–C(15) |
1.308(4) |
1.314 |
| C(17)–N(5) |
1.354(2) |
1.386 |
S(2)–C(21) |
1.729(3) |
1.805 |
S(2)–C(17) |
1.739(3) |
1.808 |
| C(17)–N(6) |
1.323(2) |
1.338 |
S(1)–C(1) |
1.717(3) |
1.789 |
S(1)–C(1) |
1.720(3) |
1.789 |
| C(17)–S(2) |
1.674(2) |
1.730 |
N(2)–C(6) |
1.306(4) |
1.339 |
N(2)–C(2) |
1.308(3) |
1.335 |
| N(1)–C(1)–S(1) |
120.5(1) |
120.1 |
N(3)–Ni(1)–N(4) |
93.5(1) |
92.0 |
N(3)–Cu(1)–N(4) |
92.22(9) |
91.9 |
| C(2)–N(3)–H(3) |
116.3(1) |
113.2 |
N(4)–Ni(1)–S(2) |
87.48(2) |
86.7 |
N(4)–Cu(1)–S(2) |
86.67(7) |
85.7 |
| C(15)–N(4)–N(5) |
118.5(1) |
119.6 |
S(2)–Ni(1)–S(1) |
85.65(3) |
88.9 |
S(2)–Cu(1)–S(1) |
89.58(3) |
90.5 |
| N(6)–C(17)–S(2) |
122.8(1) |
125.7 |
N(3)–Ni(1)–S(1) |
95.32(7) |
92.4 |
N(3)–Cu(1)–S(1) |
94.83(7) |
92.4 |
| N(5)–C(17)–S(2) |
120.6(1) |
118.5 |
N(3)–Ni(1)–S(2) |
168.47(8) |
173.0 |
N(3)–Cu(1)–S(2) |
166.17(7) |
169.8 |
| C(9)–N(3)–H(3) |
116.3(1) |
116.6 |
N(4)–Ni(1)–S(1) |
167.35(8) |
175.3 |
N(4)–Cu(1)–S(1) |
164.80(7) |
174.2 |
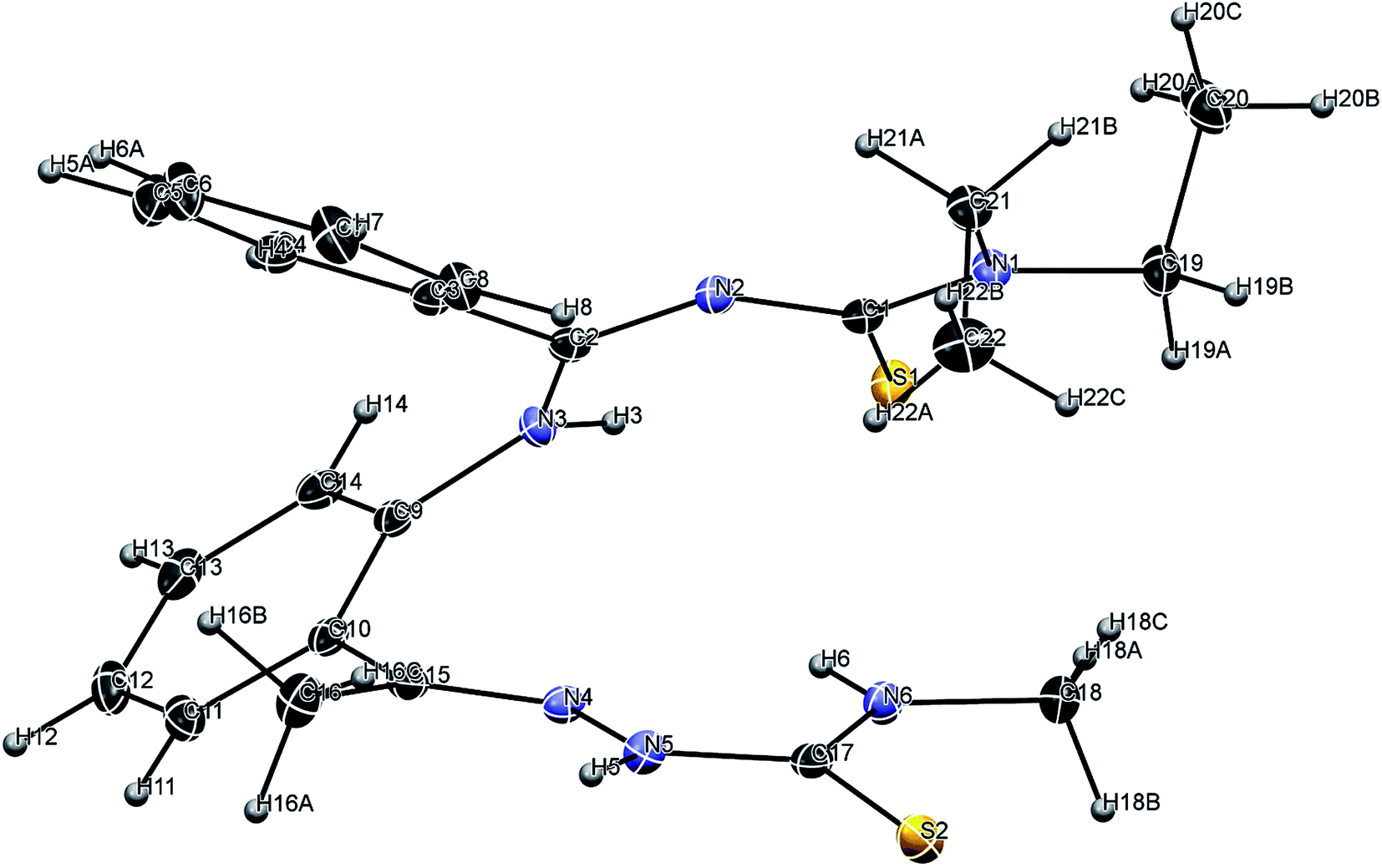
Fig. 1 depicts the molecular structure of H2L. H2L crystallized in the triclinic Pi space group. Selected bond lengths and angles are summarized in Table 2. The protonation of nitrogen atoms N3 and N5 is experimentally justified by the detection of peaks of electron density in the final Fourier map, which can be assigned to corresponding hydrogen atoms that are involved in intermolecular hydrogen bonding. The N(3)–H(3)–S(1) hydrogen bond forms an intramolecular ring of type S11(6). The C–N and CN bond distances (see Table 2) found in H2L agree well with those reported for other similar ligands containing C–N/CN bonds. The thione form is confirmed by the bond lengths of C(1)–S(1) (1.704 Å) and C(17)–S(2) (1.674 Å), which are very close to a formal CS bond length (1.60 Å). Examination of the packing diagram of H2L revealed that the structure exhibits intermolecular π–π stacking interactions between the parallel C(9)–C(14) ring and its equivalent obtained by a (−x, 1 − y, −z) symmetry transformation, although the geometrical parameters with a distance of 3.575 Å and a shift equal to 1.440 Å indicate a rather weak interaction, as shown in Fig. S8.†
 |
| | Fig. 1 Perspective view (25% probability ellipsoids) of H2L with the atom numbering scheme. | |
A perspective view of complexes 1 and 2 with the atomic numbering schemes is depicted in Fig. 2 and 3, while selected bond lengths and angles are given in Table 2. Complexes 1 and 2 crystallized in the triclinic Pbca space group. The metal ion adopted a square planar geometry with H2L ligands bonded as dibasic tetradentate (SNNS) donors. The trans angles are N(3)–Ni(1)–S(2), 168.47(8)° and N(4)–Ni(1)–S(1), 167.35(8)° (for 1) and N(3)–Cu(1)–S(2), 166.17(7)° and N(4)–Cu(1)–S(1), 164.80(7)° (for 2), which showed a deviation from the expected linear trans geometry, suggesting distortion in the square-planar coordination geometry. The metal bond distances Ni–N and Ni–S in complex 1 and Cu–N and Cu–S in complex 2 (Table 2) are in good agreement with reported nickel(II) and copper(II) complexes.31a,b Furthermore, there is intermolecular hydrogen bonding between the atoms (C–H⋯N/S contact) of H2L and its complexes, 1 and 2, as shown in Table 3. The unit cell packing diagram of the compounds, along with hydrogen bonding, is given in Fig. S9.†
 |
| | Fig. 2 Perspective view (25% probability ellipsoids) of complex 1 with the atom numbering scheme. | |
 |
| | Fig. 3 Perspective view (25% probability ellipsoids) of complex 2 with the atom numbering scheme. | |
Table 3 Hydrogen bonds for H2L and its complexes, 1 and 2a [Å and °]
| Compound |
D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
∠(DHA) |
| Symmetry operation: H2L: ‘x, y, z’; ‘−x, −y, −z’, complex 1: ‘x, y, z’; ‘−x + 1/2, −y, z + 1/2’; ‘x + 1/2, −y + 1/2, −z’; ‘−x, y + 1/2, −z + 1/2’; ‘−x, −y, −z’; ‘x − 1/2, y, −z − 1/2’; ‘−x − 1/2, y − 1/2, z’; ‘x, −y − 1/2, z − 1/2’, complex 2: ‘x, y, z’; ‘−x + 1/2, −y, z + 1/2’; ‘x + 1/2, −y + 1/2, −z’; ‘−x, y + 1/2, −z + 1/2’; ‘−x, −y, −z’; ‘x − 1/2, y, −z − 1/2’; ‘−x − 1/2, y − 1/2, z’; ‘x, −y − 1/2, z − 1/2’ [D = donator, A = acceptor]. |
| H2L |
N(3)–H(3)–S(1) |
0.86 |
2.28 |
2.968(2) |
137.2 |
| N(5)–H(5)–S(2) |
0.86 |
2.61 |
3.351(2) |
145.2 |
| N(6)–H(6)–N(4) |
0.86 |
2.19 |
2.577(2) |
106.9 |
| 1 |
C(2)–H(2A)–S(1) |
0.97 |
2.57 |
2.963(3) |
104.3 |
| C(20)–H(20A)–N(5) |
0.96 |
2.24 |
2.600(5) |
101.2 |
| 2 |
C(16)–H(16B)–N(5) |
0.96 |
2.21 |
2.611(4) |
103.9 |
| C(19)–H(19B)–S(1) |
0.97 |
2.56 |
2.949(3) |
104.2 |
Theoretical calculations
HOMO and LUMO are referred to as frontier orbitals. The energy difference between the HOMO and LUMO is termed as the HOMO–LUMO energy gap. The difference in energy between two frontier orbitals can be used to predict the strength and stability of transition metal complexes as well as the colors they produce in solution. Calculations using the density functional theory (DFT) method of GAUSSIAN-09 were carried out to determine the electronic structures of the compounds. A molecule with a small gap is more polarized and is known as a soft molecule.32 Lowering of the HOMO–LUMO band gap is essentially a consequence of stabilization of the LUMO due to the strong electron-accepting ability of an electron-acceptor group.
The molecular structure of H2L and the deprotonated forms of its complexes 1 and 2 were characterized through computational studies of the solid phase. Contour plots of frontier molecular orbitals of the complexes and the energy gap were also calculated (Fig. 4). Positive and negative regions are shown in red and blue, respectively, and the geometrical parameters have already been given in Table 1. In general, there is good correlation between the optimized structures and the X-ray crystal structures of H2L and its complexes 1 and 2. The perfect square planar geometries of the complexes are also reflected in their calculated structures. It is observed that in the complexes, the electron density of the HOMO is localized largely on the atoms (N and S) surrounding the metal ion and to a much smaller extent on the ligand, whereas in the LUMO, major contributions are from the ligand and to a lesser extent from the atoms surrounding the metal ion. Therefore, electron transfer occurs from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). It can be inferred that electron transfer in complexes is related to LMCT. The calculated HOMO energies of the complexes vary as H2L (−5.68 eV) < 1 (−5.25 eV) < 2 (−5.31 eV) and those of the LUMO exhibit a similar trend: H2L (−2.04 eV) < 1 (−2.15 eV) < 2 (−2.05 eV). The HOMO–LUMO energy gaps in H2L (3.64 eV), complex 1 (−3.12 eV) and complex 2 (−3.20 eV) are only slightly different and their order is consistent with the experimentally observed order of the biological efficiencies of the complexes. The calculated atomic charges and electronic configurations of donor atoms and metal-centered atoms are listed in Table 4.
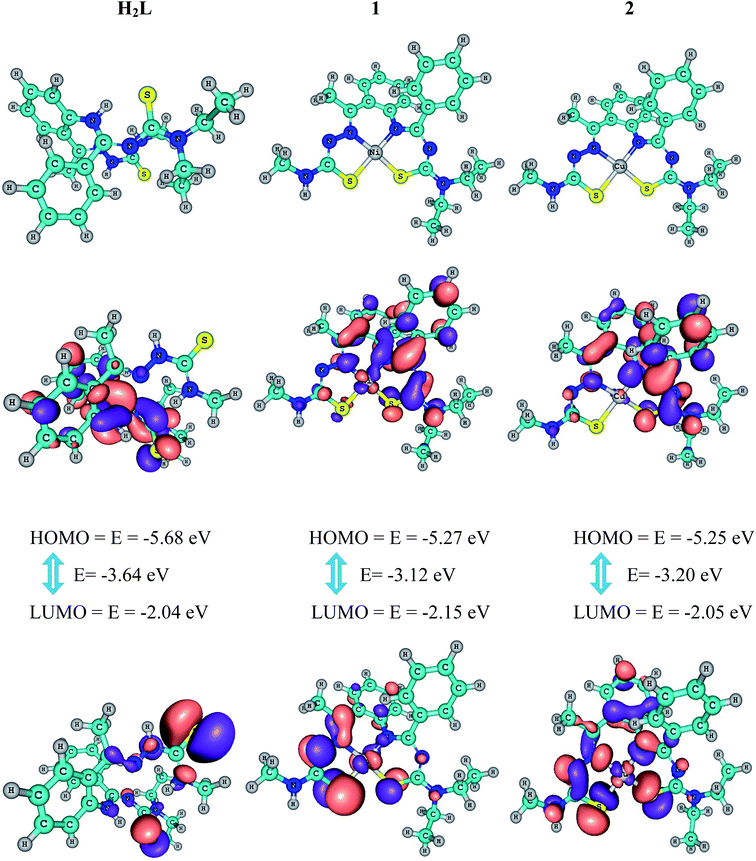 |
| | Fig. 4 Frontier molecular orbitals of compounds, their HOMO–LUMO energy gap and their B3LYP/6-311G ground state optimized geometry (top). | |
Table 4 Charges (a.u.) and electronic configurations of atoms for complexes 1 and 2
| Atom |
Complex 1 |
Complex 2 |
| Charge (eV) |
Electronic configuration |
Charge (eV) |
Electronic configuration |
| Ni/Cu |
0.95397 |
[Core]4S(0.41)3d(8.60)4p(0.02)5S(0.02) |
1.19451 |
[Core]4S(0.43)3d(9.35)4p(0.01)5S(0.01) |
| N1(6) |
−0.65053 |
[Core]2S(1.32)2p(4.31)3p(0.01) |
−0.72124 |
[Core]2S(1.33)2p(4.37)3p(0.01) |
| N2(7) |
−0.37386 |
[Core]2S(1.35)2p(4.00) 3p(0.02) |
−0.42138 |
[Core]2S(1.36)2p(4.04)3p(0.02) |
| S1(2) |
−0.28649 |
[Core]3S(1.80)3p(4.47)4S(0.01)4p(0.01) |
−0.34276 |
[Core]3S(1.81)3p(4.52)4S(0.01)4p(0.01) |
| S2(3) |
−0.36154 |
[Core]3S(1.81)3p(4.53)4p(0.01) |
−0.42362 |
[Core]3S(1.82)3p(4.58)4S(0.01)4p(0.01) |
| C1(11) |
0.34264 |
[Core]2S(0.86)2p(2.77)3p(0.02) |
0.34451 |
[Core]2S(0.86)2p(2.77)3p(0.02) |
| C2(39) |
0.31027 |
[Core]2S(0.86)2p(2.79)3S(0.01)3p(0.03) |
0.30966 |
[Core]2S(0.87)2p(2.79)3S(0.01)3p(0.03) |
From Table 4, it can be observed that the electron populations on the s and p orbitals of nitrogen and sulfur donor atoms in both complexes are less than the expected values of valence orbitals, whereas the computed electron population in the central ion in both complexes 1 and 2 is more than the expected value in M2+ (Ni, Cu) with d8 and d9 electronic configurations, respectively.33 The calculated formal charges on the metal ions (1 = 0.954 eV; 2 = 1.195 eV) in the complexes were lower than the formal charge of +2, confirming a significant charge donation from the ligand, H2L. This confirms electron transmission from donor atoms toward the central metal. The calculated electronic configurations (Table 4) of the donor atoms with reference to s and p orbitals are consistent with electron donation towards the metal ion.
DNA interaction studies
Stability of compounds. Before carrying out DNA binding studies, the stability of the complexes is paramount with respect to their application. The stability of the selected compounds (H2L, 1 and 2) has been determined in Tris–HCl buffer (5 mM Tris–HCl-50 mM NaCl buffer, pH = 7.2) at four different time points (0 min, 1 h, 6 h and 12 h) using the UV absorption method. The UV-vis spectral results did not reveal any appreciable change in either the intensity or the position of the absorption bands in Tris–HCl buffer solution (Fig. S3†). This demonstrates that the compounds are stable under physiological conditions.
Fluorescence emission titration. DNA is often a vital target to mediate cell apoptosis or necrosis. Therefore, the binding affinity of the metal complexes to CT-DNA was used as a primary criterion for assessing their suitability as chemotherapeutic agents. These studies were carried out using different spectral methods. As a basic testing method, fluorescence spectroscopy is one of the most commonly-used techniques for exploring the interaction of any drug with CT-DNA.34 This technique is commonly used to study the binding mode (and its nature) between DNA and metal complexes. Emissive titration experiments were performed by monitoring the changes in emission intensity as a result of aliquot addition of DNA. Usually, intercalative modes of interaction between metal complexes and DNA result in hypochromism with or without red/blue shift; on the other hand, non-intercalative/electrostatic interaction causes hyperchromism. Therefore, typical emissive titration curves for the selected compounds in the absence and presence of CT-DNA at different increasing concentrations were implemented.From the emissive titration spectra (Fig. 5), it is apparent that the addition of an increasing concentration of CT-DNA (0–50 μM) to a solution of H2L resulted in hypochromism (2.41%) at 304 nm along with a small red shift (∼2 nm). Complexes 1 and 2 show different behavior relative to that of the ligand and exhibit bands at 406 and 528 nm, showing hypochromism of about 36.36% and 53.27%, respectively, with a small red shift (∼4 nm). The observed hypochromism is due to an intercalative mode of binding involving a strong stacking interaction between the extended aromaticity of the ligand and the base pairs of DNA.35 The binding of compounds to DNA leads to hypochromism, which provides a measure of the strength of the intercalative binding. Based on the emission enhancement, the intrinsic binding constant was obtained according to the Scatchard equation:
| Cb = Ct(F – F0)/(Fmax − F0), |
where
Ct is the total compound concentration,
F is the observed fluorescence emission intensity at a given DNA concentration,
F0 is the intensity in the absence of DNA and
Fmax is the fluorescence of the totally bound compound. The fluorescence spectra of the tested compounds with an increasing concentration of CT-DNA are depicted in
Fig. 5. Intrinsic binding constants (
Kb) were cast in the form of a Scatchard plot of
r/
Cf versus r, where
r is the binding ratio of
Cb/[DNA] and
Cf is the free ligand concentration. The intrinsic binding constants (
Kb) for the interaction of
H2L,
1 and
2 with CT-DNA are listed in
Table 5. The binding constants (
Kb) of the compounds were in the range of 4.30 × 10
3 to 8.50 × 10
4 M
−1. However, the complexes interact with CT-DNA more strongly than the ligand does. The observed values of
Kb revealed that the compounds bind to DNA
via an intercalative mode, where the order of DNA-binding affinity is
1 >
2 >
H2L. However, the binding mode needs to be confirmed through some more experiments.
 |
| | Fig. 5 Fluorescence titrations of H2L, 1 and 2 (25 μM) with CT-DNA (0–50 μM) (left). Scatchard plots of r/Cf vs. r for the selected compounds (right). | |
Table 5 Fluorescence spectral parameters for compounds H2L, 1 and 2 bound to CT-DNA
| Compound |
Kb |
Ksv |
Kapp |
| H2L |
4.30 × 103 |
9.3 × 103 |
8.01 × 103 |
| 1 |
8.50 × 104 |
1.82 × 104 |
3.21 × 105 |
| 2 |
4.49 × 104 |
2.17 × 104 |
3.45 × 105 |
Ethidium bromide displacement study. Fluorescence-quenching experiments with EB-bound DNA were undertaken to understand the mode of DNA interaction with the metal complexes. The molecular fluorophore, EB, emits intense fluorescence at about 600 nm in the presence of DNA due to its strong intercalation with adjacent DNA base pairs.36 Addition of a second molecule, which binds to DNA more strongly than EB, (the fluorescence intensity of EB increases by almost 20-fold after binding to DNA) would displace the bound EB and quench the DNA-induced EB emission. The extent of quenching of the fluorescence of the EB bound to DNA would reflect the extent of DNA binding of the second molecule.37 Keeping the abovementioned fact in mind, as the concentration of H2L and its complexes, 1 and 2, increased, the emission band at 604 nm (Fig. 6) for EB exhibited hypochromism up to 26.97%, 47.95% and 31.41%, respectively, with a slight red/blue shift from the initial fluorescence. The quenching of the emission intensity of ethidium bromide on addition of these compounds shows that the complexes probably compete with EB for binding with DNA. Further quantitative assessment of the magnitude of interaction was ascertained by the classical Stern–Volmer equation:
where I0 and I are the emission intensities of EB-bound CT-DNA in the absence and presence of quencher (complexes) concentration [Q], respectively, which gave the Stern–Volmer quenching constant (Ksv). Ksv was obtained from the slope of a plot of I0/I versus [Q], as shown in Fig. 6. The quenching constant (Ksv) values obtained indicated that the complexes strongly bind to DNA. These are listed in Table 5.
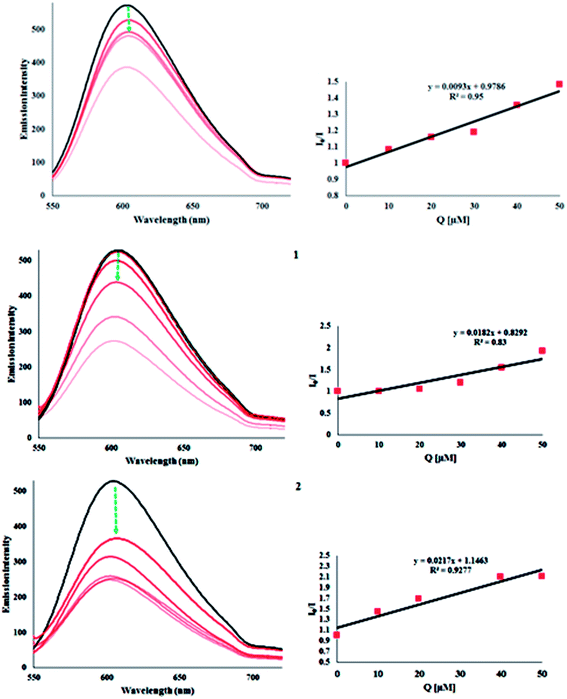 |
| | Fig. 6 Fluorescence titrations of H2L, 1 and 2 (0–50 μM) with EB-bound CT-DNA (7.5 μM) (left). Scatchard plots of I0/I vs. Q for selected compounds (right). | |
Furthermore, the apparent DNA binding constants (Kapp) were also calculated using the following equation:
where
KEB = 1.0 × 10
−7 M
−1 is the DNA binding constant of EB, [EB] is the concentration of EB (7.5 μM) and [M
50%] is the concentration of the compound used to obtain a 50% reduction in the fluorescence intensity of DNA pretreated with EB. The
Kapp values for the ligand and complexes
1 and
2 are given in
Table 5. The DNA binding ability of the compounds follows the order
1 ≈
2 >
H2L, which is consistent with the results, obtained from the abovementioned fluorescence spectral studies.
Viscosity measurement. This technique gives us useful information on how the conformation of DNA is influenced by the binding mode of the metal complex to DNA. In general, the viscosity of CT-DNA increases when a compound binds with DNA in intercalating mode but remains unchanged when a compound binds with DNA in electrostatic mode.38 When H2L and its complexes, 1 and 2, were treated with CT-DNA (200 μM), as the concentrations of the compounds (0–120 μM) increased from a ratio of r = 0–0.1(1/r = [compound]/[DNA]), the relative viscosity of DNA enlarged steadily in the order 2 > 1 > H2L. The relative viscosity of DNA solution increases upon addition of all the compounds and it is more pronounced in the case of the complexes (Fig. 7). The observed behavior of the DNA viscosity upon addition of the compounds may be considered as evidence of the existence of an intercalative binding mode to DNA, a conclusion that clarifies the preliminary indications derived from fluorescence spectroscopy studies.
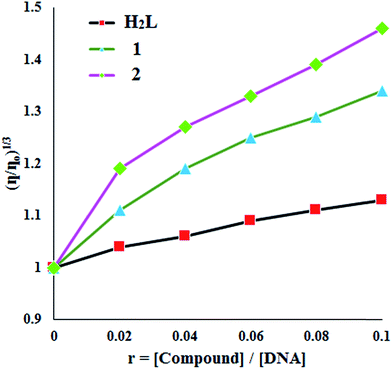 |
| | Fig. 7 Relative viscosity (η/η0)1/3 of CT-DNA in Tris–HCl buffer solution in the presence of tested compounds at increasing concentrations (r = 0–0.1). | |
BSA interaction studies
Fluorescence spectral study. Interactions between complexes and the most abundant blood protein, bovine serum albumin, have attracted immense current interest because of its structural homology with human serum albumin.39a To understand the mechanism of interactions between the selected compounds and BSA, fluorescence quenching experiments have been carried out. The fluorescence spectra of BSA showed a broad band with a maximum at ∼340 nm. The relative ratios of fluorescence intensity for three amino acids and their functions were reported earlier.33 Therefore, the fluorescence behavior of BSA can provide significant information about its structure, dynamics, and protein folding. A solution of BSA (1 μM) was titrated with various increasing concentrations of compounds H2L, 1 and 2 (0–50 μM) in the wavelength range of 290–430 nm (λex = 280 nm). Fig. 8 shows the effect of increasing concentration of compounds on the fluorescence emission of BSA.
 |
| | Fig. 8 Fluorescence titrations of H2L, 1 and 2 (0–50 μM) with BSA (1 μM) (left). Stern–Volmer plots of I0/I vs. Q and Scatchard plots of log[I0 − I/I] vs. log[Q] for selected compounds (right). | |
The extent of quenching of fluorescence intensity, as expressed by the Stern–Volmer constant (Ksv), is a measure of the protein binding affinity of the complexes. To gain an insight into the quenching progression, the quenching constant (Kq) was evaluated from the following Stern–Volmer equation:
| I0/I = 1 + Ksv[Q] = 1 + Kqτo[Q] |
where
I0 and
I are the fluorescence intensities in the absence and presence of the quencher, respectively,
Kq is the quenching rate constant,
τo is the fluorescence life time of biopolymer BSA (
τo = 10
−8 s),
39b and
Ksv and [Q] are the Stern–Volmer quenching constant and the concentration of the quencher, respectively. The quenching constant (
Kq) was obtained from a plot of log(
I0 −
I)/
I versus log[Q] (
Fig. 8). It can be observed that the
Kq value of BSA is higher than 1.0 × 10
12, which is much higher than the molecular fluorescence diffusing constant, as shown in
Table 6. Therefore, the fluorescence quenching mechanism of the compounds-BSA system cannot be dynamic quenching. It may be static quenching. Nevertheless, the hypothesis needs to be proved through some more experiments.
Table 6 Quenching parameters of BSA for compounds H2L, 1 and 2
| Compound |
Ksv |
Kq |
Kbin |
‘n’ |
| H2L |
1.83 × 105 |
1.83 × 1013 |
6.05 × 107 |
1.5 |
| 1 |
1.19 × 105 |
1.19 × 1013 |
1.34 × 109 |
1.9 |
| 2 |
1.02 × 105 |
1.02 × 1013 |
1.44 × 109 |
1.8 |
UV-visible spectral study. It has been proved that the fluorescence quenching mechanism of the compound is static quenching by UV-vis absorption spectral studies. The fluorescence quenching mechanisms are usually classified as either static or dynamic quenching. Static quenching usually results from the formation of a complex between the quencher and the fluorophore in the ground state, whereas in dynamic quenching the fluorophore and the quencher come into contact with each other during the transient existence of the excited state.40 UV-visible absorption spectroscopy is an effective method of identifying the type of quenching. UV-visible spectra of BSA in the absence and presence of the compounds (Fig. 9) showed that the absorption intensity of BSA was enhanced as the complexes were added and there was a small blue shift. It was also revealed that there existed a static interaction between BSA and the added complexes due to the formation of a ground state complex of the type BSA-compound.
 |
| | Fig. 9 Absorbance spectra of the compounds H2L, 1 and 2 with BSA. | |
Binding constant and binding site number. The binding constant Kbin was calculated using the Scatchard equation
| log[I0 − I/I] = logKbin + nlog[Q], |
where Kbin is the binding constant of the compound with BSA and n is the number of binding sites. The number of binding sites (n) and the binding constant (Kbin) have been found from the plot of log(I0 − I)/I versus log[Q] (Fig. 8).41 The calculated Kbin and n values are given in Table 6. The calculated value of n is around 2, indicating the existence of two binding sites in BSA for all the compounds. The higher values of Kq and Kbin indicate a strong interaction between the BSA protein and the complexes compared to the ligand used in this study. It is noted that the binding constants of the compounds follow the order 2 > 1 > H2L, which is consistent with the results obtained from the abovementioned binding affinity studies.
Catechol oxidase activity
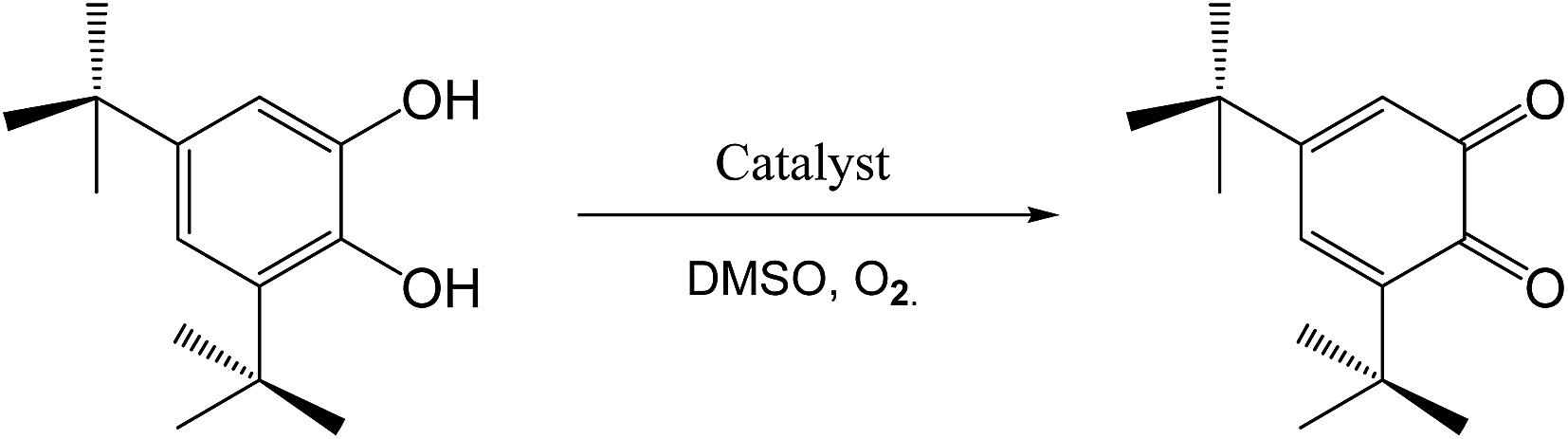
3,5-Di-tert-butylcatechol (3,5-DTBC) was employed as a substrate to study the catalytic performance of H2L and its complexes, 1 and 2, mainly because its low reduction potential makes it easy to oxidize and the bulky tert-butyl groups prevent further oxidation such as ring opening and it has shown a low quinone–catechol reduction potential (Scheme 4).42 The reactions were carried out at room temperature in aerobic conditions and were monitored by fluorescence spectroscopy. Moreover, the oxidation product of 3,5-di-tert-butylbenzoquinone (3,5-DTBQ) is highly stable and exhibited a characteristic emission intensity at λemis = 437 nm (λex = 401 nm) in pure DMSO. To monitor the reaction, 1 × 10−4 M solutions of the selected compounds were treated with 100 equivalents of 3,5-DTBC. Upon addition of the catecholic substrate, a new band (Fig. 10) gradually appeared at about 437 nm due to the formation of the oxidized product, 3,5-DTBQ. Thus, the experiment clearly proves that the oxidation of 3,5-DTBC to 3,5-DTBQ is catalyzed by the synthesized compounds, as it is well established that 3,5-DTBQ shows a maximum at λemis = 437 nm in pure DMSO.
 |
| | Scheme 4 Catalytic oxidation of 3,5-DTBC to 3,5-DTBQ in DMSO in the presence of oxygen. | |
 |
| | Fig. 10 Oxidation of 3,5-DTBC by H2L and complexes 1 and 2, monitored by fluorescence spectroscopy (left). Lineweaver–Burk plot for tested compounds (right). | |
Kinetic study of the catecholase activity
To understand the kinetic aspects of catalysis for the tested compounds, the rate constants for the compounds as catalysts were determined by traditional initial rate methods (detailed description provided in the Experimental section). The catalytic behavior shows saturation kinetics and a treatment based on the Michaelis–Menten model seemed to be appropriate under excess substrate conditions.43a The observed rate constants (ki) were extracted by the initial rate method. Plots of ki vs. [3,5-DTBC] gave non-linear curves with decreasing slopes (Fig. S10†), which are best described by the following equations.
A plausible mechanism for 3,5-DTBC oxidation promoted by metal(II) complexes is depicted in Scheme 5. Unfortunately, we were unable to characterize the intermediates. However, when a mixture of starch and potassium iodide solution was added to a mixture of complex and 3,5-DTBC, a blue coloration developed, indicating that hydrogen peroxide was produced during the course of the reaction. It is interesting to note that no blue coloration was observed in the absence of 3,5-DTBC. A believable mechanistic path for the formation of H2O2 as a by-product during the oxidation of 3,5-DTBC to 3,5-DTBQ, catalyzed by metal(II) compounds, was suggested by Chyn and Urbach.43b
 |
| | Scheme 5 Plausible mechanism for oxidation of 3,5-DTBC by complexes 1 and 2. | |
According to the reported plausible mechanism for the generalized catecholase reaction (Scheme 5), electron transfer is mainly assisted by the metal center and then further delocalized via the CN bond of metal(II) complexes to the adjacent conjugate system. Particularly, the chelate formation of complexes extended by aromatic system containing delocalised π-electron cloud and the probable thione–thiol tautomerism showed in Scheme 3. The Michaelis–Menten constant (Km) and the maximum initial rate (Vmax) were determined by linearization using Lineweaver–Burk plots (Fig. 10). The rate constants for dissociation of substrates S (i.e. turnover number, kcat) were calculated from the graphs of 1/V vs. 1/[S] (Fig. 10), known as the Lineweaver–Burk graph, using the abovementioned equation and all these parameters are listed in Table 7. Upon comparison of the values in Table 7, it may be stated that complexes 1 and 2 are highly efficient catalysts, wherein the order of their activity is 2 > 1 > H2L.
Table 7 Kinetic parameters for the catecholase activity of H2L and complexes 1 and 2
| Catalyst |
Km (M) |
Vmax (Mm−1) |
Kcat (h−1) |
| H2L |
9.16 × 10−3 |
3.64 × 10−4 |
91.3 |
| 1 |
1.12 × 10−2 |
5.25 × 10−4 |
116 |
| 2 |
1.46 × 10−2 |
9.17 × 10−4 |
146 |
Phosphate hydrolysis activity
Many metalloenzymes with hydrolytic properties contain metal ions in close proximity to the active site. The selected compounds meet the prerequisites for use as a functional model system for hydrolytic enzymes. To study the phosphatase activity of compounds, 4-nitrophenyl phosphate disodium salt hexahydrate (4-NPP) (Scheme 6) was the preferred choice of substrate.44 Its hydrolytic tendency was detected by monitoring the time evolution of the p-nitrophenolate ion (4-NP) in DMSO using emission intensities at λemis = 436 nm (λex = 401 nm) over a wavelength scan of 400–800 nm over 2 hours with 40 equivalents of the substrate being used relative to the catalyst.
 |
| | Scheme 6 Catalytic hydrolysis of 4-nitrophenyl phosphate disodium salt hexahydrate (4-NPP) to the 4-nitrophenolate ion (4-NP) in DMSO. | |
Kinetic study of the phosphatase activity
Kinetic studies of compounds were carried out using the initial-slope method by monitoring the rate of the increase in the emissive intensity band at 423 nm, which corresponds to the increase in the concentration of 4-nitrophenolate.45 The catalytic behavior shows saturation kinetics and a treatment based on the Michaelis–Menten model seemed to be appropriate under excess substrate conditions.46 The observed rate constants (ki) were extracted by the initial rate method. Plots of ki vs. [4-NPP] gave non-linear curves with decreasing slopes (Fig. S11†), which are best described by the kinetics of the catecholase equation. Understandably, the reaction displays first-order kinetics at lower concentrations and gradually differs from unity at higher concentrations. The Michaelis–Menten constant (Km) and the maximum initial rate (Vmax) were determined by linearization using Lineweaver–Burk plots (Fig. 11). The rate constants for dissociation of substrates S (i.e. turnover number, kcat) were calculated from the graphs of 1/V vs. 1/[S] (Fig. 11), known as Lineweaver–Burk graphs, using the kinetics equation; all these parameters are listed in Table 8. The results indicate first-order rate constant values, which are comparable to the reported values for phosphate bond cleavage.47 The order of their catalytic activity is 2 > 1 > H2L.
 |
| | Fig. 11 Hydrolysis of 4-NPP by H2L and complexes 1 and 2, monitored by fluorescence spectroscopy (left). Lineweaver–Burk plots for tested compounds (right). | |
Table 8 Kinetic parameters for the phosphatase activity of H2L and complexes 1 and 2
| Catalyst |
Km (M) |
Vmax (Mm−1) |
Kcat (h−1) |
| H2L |
9.38 × 10−2 |
3.89 × 10−3 |
1876 |
| 1 |
10.2 × 10−2 |
4.53 × 10−3 |
2040 |
| 2 |
2.54 × 10−1 |
1.15 × 10−2 |
5080 |
Anticancer activity in vitro
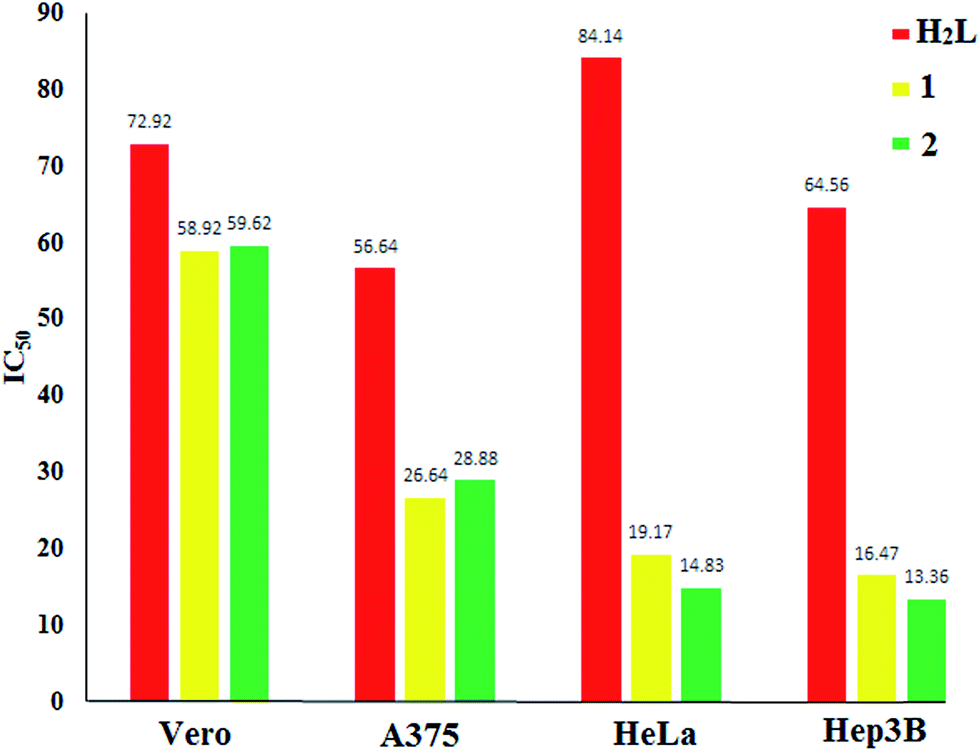
Inhibition of cancer cell viability (MTT assay). The antiproliferative activities of H2L and complexes 1 and 2 were appraised against the normal Vero cell line, the human melanoma skin cancer cell line (A375), human cervical cancer (HeLa) and the human hepatocellular carcinoma (Hep3B) cancer cell line using the MTT assay. Cisplatin was used as the reference compound to evaluate the cytotoxic activity. Cells were exposed to a broad range of drug concentrations (10–100 μg mL−1) for 24 h and cell viability was analyzed by colorimetric MTT assay.48 The results were analyzed by cell viability curves and expressed with IC50 values of H2L and its complexes, 1 and 2 (Table 9). The amount of cell proliferation significantly decreased in a dose-dependent manner on supplementation with the selected compounds (H2L, 1 and 2), as observed within 24 h of incubation with the normal Vero and cancer cell line, such as A375, HeLa and Hep3B, because thiosemicarbazones are known inhibitors of enzyme ribonucleotide diphosphate reductase (RDR).49 RDR is responsible for maintaining a balanced supply of dNTPs, which are required for DNA synthesis and repair and play an important role in cell proliferation. The ligand H2L expressed moderate cytotoxicity against normal and cancer cell lines. As can be observed, the results of MTT assays revealed that the compounds show notable activity against cancer cell lines (A375, HeLa, Hep3B) with low IC50 values (Fig. 12). Moreover, when we compare these observations, we can observe that the coordination of a metal to the ligand increased its cytotoxic activities. Upon confirmation of their excellent cytotoxicity properties, further staining assays were performed to determine their mechanism of action.
Table 9 In vitro cytotoxicity of the compounds in normal and cancer cell lines
| Compound |
IC50a (μg mL−1) |
| Vero |
A375 |
HeLa |
Hep3B |
| Fifty percent inhibitory concentration after exposure for 48 h in the MTT assay. Data from ref. 33 and 49. No data. |
| H2L |
72.92 ± 2.67 |
56.64 ± 1.69 |
84.14 ± 0.89 |
64.56 ± 1.24 |
| 1 |
58.92 ± 1.44 |
26.64 ± 2.37 |
15.17 ± 2.03 |
14.47 ± 2.31 |
| 2 |
59.62 ± 1.02 |
28.88 ± 1.85 |
12.83 ± 1.65 |
11.36 ± 1.57 |
| Cisplatinb |
NDc |
12.10 ± 0.6 |
13.00 ± 2.01 |
NDc |
 |
| | Fig. 12 IC50 values of compounds H2L, 1 and 2 on normal Vero and cancer cell lines such as A375, HeLa and Hep3B. | |
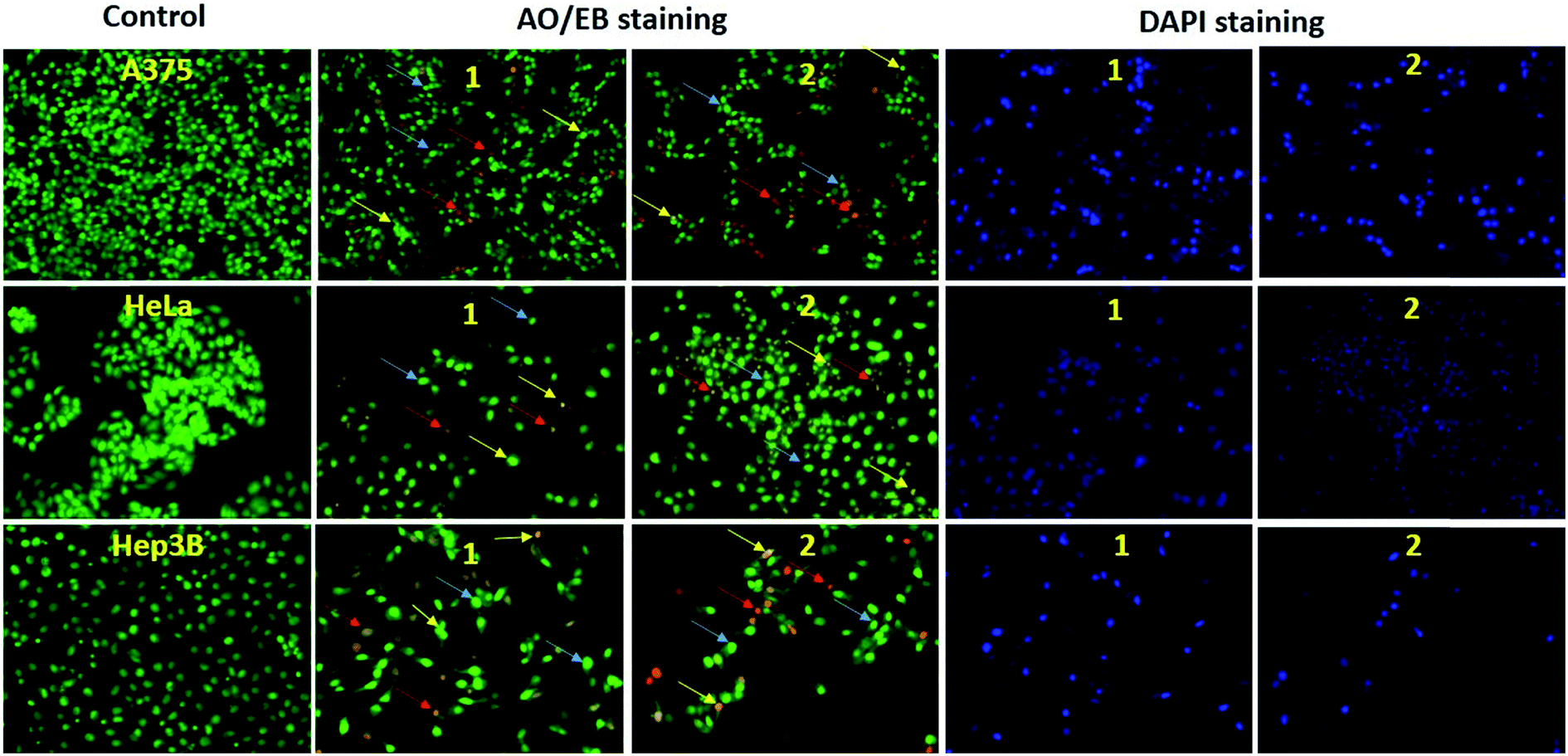
Cell morphological observation for apoptosis induction. To determine the mode of cell death, cells were grown on coverslips, treated with the compounds for 24 h (Fig. 13) and observed for changes in morphology. Apoptosis is the process of programmed cell death (PCD) that occurs in multicellular organisms. These morphological changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation and global mRNA decay.50 Cell death apoptosis in the A375, HeLa and Hep3B cell lines induced by complexes 1 and 2 was primarily studied with respect to the changes observed in the cell morphology after the cells were stained. Apoptotic cells exhibit increased plasma membrane permeability to certain fluorescent dyes, e.g. AO/EB, Hoechst, AO/PI and DAPI.51 In this study, we have used membrane-permeable AO/EB and DAPI fluorescence staining.
 |
| | Fig. 13 Images of drug-treated A375, HeLa and Hep3B cancer cells after AO/EB and DAPI staining, followed by 24 h incubation with complexes at fixed concentrations. The yellow arrows show early apoptotic cells with blebbing, blue arrows show late apoptosis and red arrows show necrotic cells. | |
Images of the control cells and treated A375, HeLa and Hep3B cancer cells are depicted in Fig. 13. In this figure, yellow arrow(s) show early apoptotic cells with membrane blebbing, which is observed at fixed doses (10 μg mL−1) of the complexes, and blue arrow(s) exhibited late apoptotic cells with chromatin aggregation, that is, highly condensed chromatin. The red arrow(s) show necrotic cells. These are the characteristic features of apoptotic cells and are quite different from those of the control cells. Overall, the results indicate that the complexes favor necrosis at a fixed lower dose, which is in good agreement with the reported toxicity results.52
Conclusion
Two new metal(II) [M = Ni2+ (1), Cu2+ (2)] complexes have been designed and synthesized using a doubly deprotonated N2S2 hybrid benzamidine–thiosemicarbazone ligand. The characterization of the compounds was accomplished by elemental analysis and various spectroscopy techniques (IR, UV-vis and 1H NMR) as well as ESI mass spectrometry. The solid state structure of all the compounds was confirmed by single-crystal X-ray crystallography. The geometry of complexes 1 and 2 revealed a square-planar arrangement around the metal ion. On the basis of DFT studies, the HOMO–LUMO energy gap explained the charge transfer interactions occurring within the molecule. The lowering of the HOMO–LUMO band gap supported the bioactive properties of the molecule. The structural parameters of the complexes, which are in good agreement with X-ray analysis, were obtained by DFT calculations. The CT-DNA binding propensities of the compounds were determined by various physico-chemical techniques. All the experimental results showed that the complexes interact strongly with CT-DNA by an intercalative binding mode. Furthermore, the results of fluorescence quenching experiments with BSA confirmed that the complexes have binding affinity in static mode. Apart from the abovementioned interactions, the compounds showed effective bio catalytic activity toward the oxidation of 3,5-DTBC and the hydrolysis of 4-NPP. The complexes showed widespread anti-tumor potency with low IC50 values tested by MTT assay against the normal Vero cell line and skin (A375), cervical (HeLa) and liver (Hep3B) cancer cell lines. The characteristics of apoptosis in cell morphology have been observed using AO/EB and DAPI staining. The results attained from the present compounds are of importance with respect to the development of metal-based agents for anti-cancer applications.
Experimental section
Materials
All chemicals used in this study were reagent grade and were used without further purification. Solvents were purified and dried according to the standard procedures. All synthetic manipulations were routinely performed under oxygen atmosphere. Doubly distilled water was used to prepare buffers. Calf thymus DNA (CT-DNA), bovine serum albumin (BSA), and 4-nitrophenyl phosphate disodium salt hexahydrate (4-NPP) were obtained from Genei, Bangalore and Himedia, India. Ethidium bromide (EB), 3,5-di-tert-butylcatechol (3,5-DTBC), and tris(hydroxymethyl) amino methane were purchased from Sigma-Aldrich and used as received. The synthesis of N,N-(diethylaminothiocarbonyl)benzimidoyl chloride was performed as a multi-step synthesis by the standard procedure of Beyer et al.10 The reported method was used as for the synthesis of primary amines such as 2-aminoacetophenone-N4-methylthiosemicarbazone,25 except that the purification was carried out by recrystallization from ethanol.
General methods
Elemental analysis (C, H, N and S) was carried out on a Vario EL III CHNS analyzer. Infrared spectra were obtained as KBr pellets using a Perkin-Elmer FT-IR spectrophotometer in the range of 4000–400 cm−1. 1H-NMR spectra were obtained on a Bruker Ultra Shield at 300 MHz using CDCl3 as solvent and TMS as an internal reference. Mass spectra for the complexes were obtained on an advanced Q-TOF micro™ mass spectrometer using an electrospray ionization probe. All MS results are given in the form: m/z, assignment. Electronic spectra were obtained on a JASCO V-570 spectrophotometer. ESR spectra were obtained on a JEOL model JES FA200 ESR spectrometer at liquid nitrogen temperature operating at X-band frequency (9.45 GHz). Geometry optimization by the density functional theory (DFT) method was performed using the B3LYP/6-311G package. Fluorescence spectral data were obtained using a JASCO FP-8200 fluorescence spectrophotometer at room temperature. Single-crystal X-ray diffraction data collection was carried out at 100(2) K and 295(2) K on a Bruker Apex-II CCD area-detector equipped with a liquid nitrogen cryostat. Melting points were checked on a technico micro heating apparatus and are uncorrected. Stock solutions of compounds (1.0 × 10−3 M in DMSO) were stored at 4 °C and the required concentrations were prepared for all experiments. All the stock solutions were used after no more than four days. Solutions of compounds were freshly prepared 1 hour prior to biochemical evaluation. Data were expressed as the mean ± the standard deviation from three independent experiments.
Synthesis of ligand (H2L)
Solid N-(diethylaminothiocarbonyl)benzimidoyl chloride (1.018 g, 4 mmol) was added to a mixture of 2-aminoacetophenone-N4-methylthiosemicarbazone (0.889 g, 4 mmol) and triethylamine (1.01 g, 10 mmol) in 10 mL absolute ethanol. The mixture was stirred for 2 h at 60 °C. The organic solvent was evaporated under reduced pressure to dryness. The residue was dissolved in 20 mL of THF and brine solution (20 mL) was added. The organic layer was separated, dried over MgSO4, filtered, and the solvent was removed in vacuo. The residue was washed with diethyl ether and dried under vacuum to give H2L as a pure yellow solid. Single crystals suitable for X-ray determination were grown by slow evaporation of an ethanol solution of H2L at room temperature.
Yield: 45% (0.616 mg). Color: yellowish orange; MP: 170 °C; micro analytical data for C22H28N6S2 required: C, 59.97; H, 6.40; N, 19.07; S, 14.55%. Found: C, 59.65; H, 6.22; N, 19.46; S, 14.12%; IR (KBr, cm−1): 3302, 3271, 3236 (NH), 1717, 1686, 1668 (CN), 838, 781 (CS). UV-vis [CHCl3, λmax, nm (ε, dm3 mol−1 cm−1)]: 238 (21202), 280 (14089). 1H NMR (300.13 MHz; CDCl3, ppm): 1.22 (t, J = 7.1 Hz, 3H, CH3), 1.24 (t, J = 7.1 Hz, 3H, CH3), 1.28 (s, 3H, CH3), 3.20 (s, 3H, NCH3), 3.91 (q, J = 7.1 Hz, 2H, NCH2), 3.87 (q, J = 7.1 Hz, 2H, NCH2), 7.16 (t, J = 7.1 Hz, 2H, Ph), 7.17–7.21 (m, 4H, Ph + C6H4), 7.36 (m, 3H, Ph + C6H4), 8.10 (s, 1H, NH), 8.47 (s, 1H, NH), 12.72 (s, 1H, NH). ESI-MS (calcd, found, m/z) = 440.63, 441.55 (M + H)+.
Synthesis of complexes 1 and 2
Ni(OAc)2·4H2O (0.2448 g, 0.001 mole) or Cu(OAc)2·4H2O (0.1998 g, 0.001 mole) was dissolved in 20 mL of MeOH:CHCl3 (1:1, v/v) and the same solution of H2L (0.4406 g, 0.001 mol) was added at room temperature. After the solution had been continuously stirred for 30 min, three drops of Et3N in CH2Cl2 (5 mL) was added. The reaction mixture was stirred for 2 h at room temperature. The reaction was monitored by thin-layer chromatography (TLC) using silica gel on aluminium sheets with a 10/90 mixture of ethyl acetate/petroleum ether as the mobile phase. After completion of reaction, the resulting solution was filtered and the filtrate was left tranquil for the slow evaporation of the mother solvent. After three days, large, black colored crystals suitable for X-ray diffraction were obtained.
[Ni(L)] (1). Yield: 65%. Color: dark green; MP: 198 °C; micro analytical data for C22H26N6NiS2 required: C, 53.13; H, 5.27; N, 16.90; S, 12.90. Found: C, 53.01; H, 5.35; N, 16.36; S, 12.52%. IR (KBr, cm−1): 3417 (NH), 1715 (CN), 1527 (CN), 1601 (C–N), 784 (CS), 761 (C–S). UV-vis [CHCl3, λmax, nm (ε, dm3 mol−1 cm−1)]: 236 (13924), 296 (8056), 367 (8132), 431 (7300). 1H NMR (300.13 MHz, CDCl3, ppm): 1.28 (t, J = 7.2 Hz, 3H, CH3), 1.32 (t, J = 7.2 Hz, 3H, CH3), 3.74 (d, J = 5.0, 3H, NCH3), 3.76 (s, 3H, NC–CH3), 3.86 (m, 1H, NCH2), 3.89 (m, 1H, NCH2), 4.06 (m, 1H, NCH2), 4.10 (m, 1H, NCH2), 4.85 (s, br, NH), 6.40 (d, J = 8.0 Hz, 1H, C6H4), 6.78 (t, J = 7.6 Hz, 1H, C6H4), 6.85 (t, J = 7.7 Hz, 1H, C6H4), 7.10 (m, 3H, Ph), 7.27 (d, J = 7.2 Hz, 2H, Ph), 7.54 (d, J = 7.9 Hz, 1H, C6H4). ESI-MS (calcd, found, m/z) = 497.31, 498.42 (M + H)+
[Cu(L)] (2). Yield: 60%. Color: dark brown; MP: 215 °C; micro analytical data for C22H26N6CuS2 required: C, 52.62; H, 5.22; N, 16.74; S, 12.77. Found: C, 53.22; H, 5.05; N, 16.36; S, 12.52%. IR (KBr, cm−1): 3390 (NH), 1700 (CN), 1547 (CN), 1572 (C–N), 790 (CS), 764 (C–S). UV-vis [CHCl3, λmax, nm (ε, dm3 mol−1 cm−1)]: 237 (13656), 298 (7800), 374 (7912), 491 (8176). ESR (X-band, 9.45 GHz, LNT): g‖ 2.078, g⊥ 2.031, gav 2.046. ESI-MS (calcd, found, m/z) = 502.06, 503.52 (M + H)+
X-ray structure determination
Suitable single crystals of H2L and complexes 1 and 2 were mounted on a glass fiber with epoxy cement. The crystals were cut into a fitting size (less than collimator cross section diameter). Crystal data was obtained with a Bruker SMART APEX 2 at 100(2) K (H2L) and a Gemini A Ultra Oxford diffraction diffractometer at 295(2) K (complexes 1 and 2). The crystal data were obtained using graphite monochromatized Cu (Kα, λ = 0.71073 Å) for H2L and Mo (Kα, λ = 0.71073 Å) for complexes 1 and 2 throughout. The data were corrected for Lorentz and polarization effects with the SMART14 suite programs and for absorption effects with SADABS.52 A data obtaining strategy using ω and φ scans at 0.5° scan technique yielded full hemispherical data with excellent intensity statistics. Structure solutions and refinements were performed using the program SHELXS-2014.53 The structures were solved by direct methods to locate the heavy atoms, followed by difference maps for the light non-hydrogen atoms. Anisotropic thermal parameters were refined for the rest of the non-hydrogen atoms. Hydrogen atoms were placed geometrically and refined isotropically. Details of the data collection and refinement are gathered in Table 1 and important bond lengths and angles for the compounds are summarized in Table 2.
Theoretical calculation (DFT)
All calculations were performed using the GAUSSIAN09 (G09) program package with the aid of the Gauss View visualization program. Full geometry optimizations of compounds were carried out using the DFT method at the Becke's three parameter hybrid exchange functional (B3LYP) level of theory while all the non-metal atoms were described using the 6-311G basis set.54 This functional has been shown to give more accurate results for all compounds. Differences between the experimental and theoretical data were determined for the solid and gas phases. Geometry optimizations have been done without any symmetry restriction with X-ray coordinates of the molecule. Frequencies of all complexes have been computed at the same level of theory to confirm that all optimized structures are at true minima, which means they have no imaginary frequencies. At the same level and basis sets, calculations of the natural electron population, natural charge for each atom and frontier molecular orbitals of the complexes have been performed by natural bond orbital (NBO) analysis on the gas phase optimized structures.55 The molecular orbital plots have been generated using the chemcraft program package (http://www.chemcraftprog.com).
DNA-binding studies
Emissive titration. All the experiments concerning the interaction of the complexes with calf thymus (CT) DNA were performed in a mixed solvent of 5% DMSO and 95% Tris–HCl buffer (5 mM Tris–HCl/50 mM NaCl buffer for pH 7.2) for all the experiments, which was stored at 4 °C for further use. Such solutions, with the aqueous buffer DNA solutions used in the studies, never exceeded 5% DMSO (v/v) in the final solution, which was needed due to the low aqueous solubility of most compounds. All studies were performed at room temperature. The solvent medium has no influence on the compounds. The CT-DNA concentration per nucleotide was determined fluorescence spectrometry by selected compounds excitation wavelength. During titration, an equal quantity of CT-DNA was added to both the complex solution and the reference solution to eliminate the intensity of CT-DNA itself and Tris–HCl buffer was subtracted through base-line correction. Emissive titration experiments were performed with a fixed concentration of H2L and complexes 1 and 2 (25 μM). While gradually increasing the concentration (0–10 μM) of DNA, the emission intensities were recorded for selected compounds in the range of 300–600 nm. Titrations were manually done using a micropipette for the addition of CT-DNA. It is noteworthy that the DNA in double distilled water does not show any luminescence.
Ethidium bromide displacement assay. Ethidium bromide displacement assays were carried out by the addition of H2L and complexes 1 and 2 to a sample solution containing EB-DNA. The spectra were obtained at an excitation wavelength of 500 nm over an emission range of 605 nm. In the fluorescence quenching spectra, the reduction in emission intensity is a measure of the binding propensity of the complex to CT-DNA. This displacement method serves as indirect evidence to identify intercalative binding modes. EB alone showed minimal fluorescence, and the fluorescence was enhanced greatly with gradual addition of CT-DNA until maximum fluorescence was achieved due to the formation of an intercalative DNA–EB adduct. Addition of increasing amounts of the compounds to the DNA–EB adduct quenched the fluorescence. Before the emission spectra were obtained, in these systems CT-DNA was pretreated with EB in the ratio [DNA]/[EB] = 1:1 ratio for 30 minutes at room temperature to fully react. Then, the titration compounds were added to this mixture of EB-DNA and the change in the fluorescence intensity was measured.
Viscosity experiment. Viscosity measurements were carried out using an Ubbelodhe viscometer maintained at a constant temperature of 30.0 °C (±0.1) in a thermostatic bath. DNA samples of approximately 200 base pairs in length were prepared by sonication to minimize complexities arising from CT-DNA flexibility. The flow time was measured three times, after 5 min of incubation, with each addition of H2L and complexes 1 and 2 and the average flow time was taken for calculation of relative viscosity. Relative viscosities for CT-DNA in the presence and absence of the compound were calculated from the relation g = (t − to)/to, where t is the observed flow time of a DNA-containing solution and to is the flow time of Tris–HCl buffer alone. Data were presented as (η/η0)1/3 versus binding ratio (R = [compounds]/[DNA] = 0.0–0.1), where η is the viscosity of CT-DNA in the presence of the compound and η0 is the viscosity of CT-DNA alone.
Protein binding studies
Quenching of the tryptophan residues of BSA was performed using H2L and complexes 1 and 2 as quenchers. The fluorescence spectra were obtained at room temperature, with the excitation wavelength of BSA at 280 nm and the emission at 344 nm, keeping the concentration of BSA constant (1 μM) while increasing the complex concentration (0–50 μM) regularly. The excitation and emission slit widths (each 5 nm) remained constant for all the experiments. A scan rate of 500 nm min−1 was used. In addition, absorption titration experiments were carried out while keeping the concentration of the complexes (20 μM) and the BSA concentration (1 μM) constant. Titrations were manually done using a micropipette for the addition of test compounds. The Stern–Volmer and Scatchard equations and graphs were often used to study the interaction of the quencher with BSA.
Kinetic assays
Catechol oxidation. All kinetic experiments were carried out under pseudo-first-order conditions,56 with H2L and complexes 1 and 2 as the minor component. Quenching of the emission intensity of 3,5-DTBC at λemis = 437 nm (λex = 401) was monitored using the selected compounds. 100 equivalents of 1 × 10−3 M solutions of 3,5-di-tert-butylcatechol (3,5-DTBC) in DMSO were added to 1 × 10−4 M solutions of the selected compounds in DMSO under aerobic conditions. The emissive intensity of the resultant reaction mixture was plotted with respect to wavelength at regular intervals of 15 min with a spectrophotometer in the range of 400–700 nm.7 The dependence of the rate on various concentration and different kinetic parameters were obtained by treatment of 1 × 10−4 M solution of compounds with 20 to 500 equivalents of substrate and monitoring the upsurge in emission at 437 nm (the peak corresponding to the quinone band maxima) as a function of time.
Phosphate ester hydrolysis. Many metalloenzymes with hydrolytic properties contain metal ions in close proximity to the active site. The selected compound meets the prerequisites for use as a functional model system for hydrolytic enzymes. The disodium salt of 4-nitrophenylphosphate (4-NPP) was the preferred choice of substrate. Its hydrolytic tendency was detected spectrophotometrically by monitoring the time evolution of p-nitrophenolate57 in DMSO (λmax = 436 nm) through a wavelength scan from 400 to 600 nm over 2 hours. Quenching of the emission intensity of 4-NPP at 437 nm (excitation wavelength at 401 nm) was monitored using H2L and complexes 1 and 2. Measuring the hydrolase activities involved the preparation of stock solutions of compounds (0.05 × 10−3 M) and the substrate 4-NPP (1 × 10−3 M), at higher concentrations in pure DMSO. The dependence of the rate on various concentration and different kinetic parameters were obtained by treatment of a 0.05 × 10−3 M solution of different compounds with 40 equivalents of substrate (the peak corresponding to the phenolate band maxima) as a function of time.
MTT assay
Maintenance of cancer cell lines. The A375 (human malignant melanoma skin cancer), HeLa (human cervical cancer), and Hep3B (human hepatocellular carcinoma cancer) cell lines were obtained from the National Centre for Cell Sciences Repository, University of Pune, India. Normal Vero and A375, HeLa and Hep3B cancer cells were maintained in a humidified atmosphere containing 5% CO2 at 37 °C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 units of penicillin, 100 μg mL−1 of streptomycin and 10% fetal bovine serum (FBS). Briefly, normal Vero and A375, HeLa, and Hep3B cancer cells were precultured in 96-well microtiter plates for 48 h under 5% CO2.
Preparation of samples for cell line testing. The compounds were dissolved in 0.1% DMSO (the concentration of DMSO did not exceed 0.1% v/v) to obtain 1 mM solutions. The samples were then diluted to 100 μM in PBS solution and filter-sterilized using a 0.22 μm syringe filter. This 100 μM solution in PBS was further used in cell cytotoxicity studies. The cells (1 × 106 cells per mL per well) were seeded in a 96-well plate. One day after seeding, the cells were treated with or without different concentrations of test compounds and re-incubated at 37 °C in a CO2 incubator for 24 h. After incubation, the cells were visualized using an inverted Olympus microscope.
Protocol for MTT assay. The effect of the selected compounds on the viability of Vero and A375, HeLa and Hep3B cancer cell lines was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The selected compounds were added to the micro wells containing the cell culture at final concentrations in the range of 10–100 μg mL−1. Then, each well was loaded with 10 μL MTT solution (5 mg mL−1 in PBS, pH = 7.4) for 4 hours at 37 °C. A purple formazan crystal was dissolved in 200 μL DMSO and the cell viability was determined by calculating the absorbance of each well at 540 nm using a BIORAD ELISA plate reader.58 All data were representative of three independent experiments and the percentage cell viability was calculated according to the following equation.
| Inhibition rate (IR%) = OD (control) − OD (drug treated cells)/OD (control) × 100. |
The corresponding IC50 (concentration of drug that inhibits cell growth by 50%) value was determined by nonlinear regression analysis using Origin 6.0 software.
Fluorescence morphological investigation. Cellular apoptotic morphological changes, which can be detected by AO/EB and DAPI staining, were studied by fluorescence microscopy. The A375, HeLa and Hep3B cancer cells (1 × 106 in number) were cultured in 6-well plates at 37 °C in an incubator with fixed concentrations of complexes 1 and 2 (50 μM) for 24 h. The cells were harvested and washed with ice-cold phosphate-buffered saline (PBS) and 40 μL of AO/EB solution (1 part of 100 μg mL−1 of AO in PBS; 1 part of 100 μg mL−1 of EB in PBS) was added.59 After staining, the cells were washed with PBS twice, suspended in 200 μL of PBS, and the nuclear morphology was observed under a fluorescence microscope after less than 20 minutes. For DAPI staining, the treated cells were fixed with 80% ethanol at room temperature for 30 min. The fixative was removed and the cells were washed 3 times with PBS before incubating with DAPI (1 μg mL−1) for 45 min at room temperature in the dark. Both techniques were used to determine viable cells, early apoptotic cells with blebbing, and necrotic cells. Acridine orange intercalates into the DNA and gives a green fluorescence and thus the viable cells appear with a green nucleus while early apoptotic cells show condensed or fragmented nuclei. EB is taken up only by the non-viable cells giving a bright orange nucleus of the dead cells overwhelming the acridine orange stain.60 DAPI dye is effective for fixed-cell staining and quantification of DNA content. A375, HeLa and Hep3B cancer cells were mounted on a slide, and the images were observed under a fluorescent microscope using a green/blue filter with excitation at 350 nm and emission at 460 nm. At least 200 cells from each slide were counted, and the percentage of apoptotic cells was calculated on the basis of cellular morphological features. The results were shown as the mean of three independent experiments.
Acknowledgements
The authors Dr P. V. and P. V. gratefully acknowledge UGC [F. No. 40-66/2011 (SR)] for financial support. The author R. N. is thankful to DST for financial assistance (Project No. SR/FT/CS-95/2010). The authors thank The Director, CAS in Botany, School of Life Sciences, University of Madras, Chennai for providing laboratory facilities to perform the cell lines studies.
References
- T. W. Hayton and G. J. Wu, J. Am. Chem. Soc., 2008, 130, 2005–2014 CrossRef CAS PubMed.
- M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310–13313 CrossRef CAS PubMed.
-
(a) I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed;
(b) C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2008, 47, 3040–3042 CrossRef CAS PubMed.
-
(a) F. Innocenti and M. J. Ratain, Eur. J. Cancer, 2002, 38, 639–644 CrossRef CAS PubMed;
(b) W. Lee, A. C. Lockhart, R. B. Kim and M. L. Rothenberg, Oncologist, 2005, 10, 104–111 CrossRef CAS PubMed.
- S. H. V. Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef PubMed.
- J. S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- D. X. West, A. E. Liberta, S. B. Padhye, R. C. Chikate, P. B. Sonawane, A. S. Kumbhar and R. G. Xerande, Coord. Chem. Rev., 1993, 123, 49–71 CrossRef CAS.
- A. R. Cowley, J. R. Dilworth, P. S. Donnelly, E. Labisbal and A. Sousa, J. Am. Chem. Soc., 2002, 124, 5270–5271 CrossRef CAS PubMed.
-
(a) L. Beyer and R. Widera, Tetrahedron Lett., 1982, 23, 1881–1882 CrossRef CAS;
(b) L. Beyer, J. Hartung and R. Widera, Tetrahedron, 1984, 40, 405–412 CrossRef CAS.
- M. Christlieb and J. Dilworth, Chem.–Eur. J, 2006, 12, 6194–6206 CrossRef CAS PubMed.
- R. Ramachandran, G. Prakash, S. Selvamurugan, P. Viswanathamurthi, J. G. Malecki and V. Ramkumar, Dalton Trans., 2014, 43, 7889–7902 RSC.
- R. Manikandan, N. Chitrapriya, Y. J. Jang and P. Viswanathamurthi, RSC Adv., 2013, 3, 11647–11657 RSC.
- P. Anitha, N. Chitrapriya, Y. J. Jang and P. Viswanathamurthi, J. Photochem. Photobiol., B, 2013, 129, 17–26 CrossRef CAS PubMed.
-
(a) P. Vijayan, P. Viswanathamurthi, V. Silambarasan, D. Velmurugan, K. Velmurugan, R. Nandhakumar, R. J. Butcher, T. Silambarasan and R. Dhandapani, J. Organomet. Chem., 2014, 768, 163–177 CrossRef CAS;
(b) A. Winter, K. Thiel, A. Zabel, T. Klamroth, A. Poppl, A. Kelling, U. Schilde, A. Tauberta and P. Strauch, New J. Chem., 2014, 38, 1019–1030 RSC.
- F. Arnesano, M. Losacco and G. Natile, Eur. J. Inorg. Chem., 2013, 15, 2701–2711 CrossRef.
- J. D. Hoeschele, Dalton Trans., 2009, 10648–10650 CAS.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS.
-
(a) K. Das, A. Datta, P.-H. Liu, J.-H. Huang, C.-L. Hsu, W.-T. Chang, B. Machura and C. Sinha, Polyhedron, 2014, 71, 85–90 CrossRef CAS;
(b) E. Zangrando, M. T. Islamb, M. A.-A. A. A. Islam, M. C. Sheikh, M. T. H. Tarafder, R. Miyatake, R. Zahanf and M. A. Hossain, Inorg. Chim. Acta, 2015, 427, 278–284 CrossRef CAS;
(c) S. Anbu, S. Kamalraj, A. Paul, C. Jayabaskaran and A. J. L. Pombeiro, Dalton Trans., 2015, 44, 3930–3933 RSC;
(d) V. S. Li, D. Choi, Z. Wang, L. S. Jimenez, M. S. Tang and H. Kohn, J. Am. Chem. Soc., 1996, 18, 2326–2331 CrossRef.
- J. C. Venter, M. D. Adams, E. W. Myers and P. W. Li, Science, 2001, 291, 1304–1351 CrossRef CAS PubMed.
-
(a) R. Loganathan, S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Inorg. Chem., 2012, 51, 5512–5532 CrossRef CAS PubMed;
(b) R. P. Paitandi, R. K. Gupta, R. S. Singh, G. Sharma, B. Koch and D. S. Pandey, Eur. J. Med. Chem., 2014, 84, 17–29 CrossRef CAS PubMed.
- P. Jaividhya, R. Dhivya, M. A. Akbarsha and M. Palaniandavar, J. Inorg. Biochem., 2012, 114, 94–105 CrossRef CAS PubMed.
- W. Hong, H. Lee, T. H. Noh and O.-S. Jung, Dalton Trans., 2013, 42, 11092–11099 RSC.
-
(a) R. Sanyal, A. Guha, T. Ghosh, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 85–96 CrossRef CAS PubMed;
(b) M. Mitra, P. Raghavaiah and R. Ghosh, New J. Chem., 2015, 39, 200–205 RSC;
(c) M. Pait, M. Shatruk and D. Ray, Dalton Trans., 2015, 44, 11741–11754 RSC.
- D. X. West, A. A. Nassar, F. A. E. l. Saied and M. I. Ayad, Transition Met. Chem., 1999, 24, 617–621 CrossRef CAS.
- H. H. Nguyen and U. Abram, Eur. J. Inorg. Chem., 2009, 3179–3187 CrossRef CAS.
- H. H. Nguyen, J. Grewe, J. Schroer, B. Kuhn and U. Abram, Inorg. Chem., 2008, 47, 5136–5144 CrossRef CAS PubMed.
- H. H. Nguyen and U. Abram, Polyhedron, 2009, 28, 3945–3952 CrossRef CAS.
- A. Barandov and U. Abram, Inorg. Chem., 2009, 48, 8072–8074 CrossRef CAS PubMed.
- J. Schroer and U. Abram, Inorg. Chem. Commun., 2010, 13, 26–29 CrossRef CAS.
-
(a) R. Prabhakaran, P. Kalaivani, S. V. Renukadevi, R. Huang, K. Senthilkumar, R. Karvembu and K. Natarajan, Inorg. Chem., 2012, 51, 3525–3532 CrossRef CAS PubMed;
(b) R. Prabhakaran, P. Kalaivani, R. Huang, P. Poornima, V. Vijaya Padma, F. Dallemer and K. Natarajan, J. Biol. Inorg. Chem., 2013, 18, 233–247 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- P. Vijayan, P. Viswanathamurthi, P. Sugumar, M. N. Ponnuswamy, M. D. Balakumaran, P. T. Kalaichelvan, K. Velmurugan, R. Nandhakumar and R. J. Butcher, Inorg. Chem. Front., 2015, 2, 620–639 RSC.
- Q. Yu, Y. Liu, J. Zhang, F. Yang, D. Sun, D. Liu, Y. Zhou and J. Liu, Metallomics, 2013, 5, 222–231 RSC.
- A. K. Patra, T. Bhowmick, S. Ramakumar, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2008, 6966–6976 RSC.
- F. J. M. Almes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef.
- M. Alagesan, N. S. P. Bhuvanesh and N. Dharmaraj, Eur. J. Med. Chem., 2014, 78, 281–293 CrossRef CAS PubMed.
- Y. J. Liu, Z. H. Liang, Z. Z. Li, J. H. Yao and H. L. Huang, J. Organomet. Chem., 2011, 696, 2728–2735 CrossRef CAS.
-
(a) U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed;
(b) J. R. Lackowicz and G. Weber, Biochem. J., 1973, 12, 4161–4170 CrossRef.
- Y. J. Hu, Y. Ou-Yang, C. M. Dai, Y. Liu and X. H. Xiao, Biomacromolecules, 2010, 11, 106–112 CrossRef CAS PubMed.
- S. Anbu, R. Ravishankaran, A. A. Karande and M. Kandaswamy, Dalton Trans., 2012, 41, 12970–12983 RSC.
- M. U. Triller, D. Pursche, W. Y. Hsieh, V. L. Pecoraro, A. Rompel and B. Krebs, Inorg. Chem., 2003, 42, 6274–6283 CrossRef CAS PubMed.
-
(a) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS PubMed;
(b) J. P. Chyn and F. L. Urbach, Inorg. Chim. Acta, 1991, 189, 157–163 CrossRef CAS.
- T. Gajda, Y. Düpre, I. Török, J. Harmer, A. Schweiger, J. Sander, D. Kuppert and K. Hegetschweiler, Inorg. Chem., 2001, 40, 4918–4927 CrossRef CAS PubMed.
- S. C. Batista, A. Neves, A. J. Bortoluzzi, I. Vencato, R. A. Peralta, B. Szpoganicz, V. V. E. Aires, H. Terenzi and P. C. Severino, Inorg. Chem. Commun., 2003, 6, 1161–1165 CrossRef CAS.
- R. Sanyal, A. Guha, T. Ghosh, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 85–96 CrossRef CAS PubMed.
- S. E. Bunn, C. T. Liu, Z. L. Lu, A. A. Neverov and S. R. Brown, J. Am. Chem. Soc., 2007, 129, 16238–16248 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- X. Q. Zhou, Q. Sun, L. Jiang, S. Tong Li, W. Gu, J. L. Tian, X. Liu and S. P. Yan, Dalton Trans., 2015, 44, 9516–9527 RSC.
- S. S. Bhat, A. S. Kumbhar, A. A. Kumbhar and A. Khan, Chem.–Eur. J., 2012, 18, 16383–16392 CrossRef CAS PubMed.
- S. Mukhopadhyay, P. K. Panda, D. N. Das, N. Sinha, B. Behera, T. K. Maiti and S. K. Bhutia, Acta Pharmacol. Sin., 2014, 35, 814–824 CrossRef CAS PubMed.
- SMART & SAINT Software Reference manuals, version 5.0, Bruker AXS Inc., Madison, WI, 1998 Search PubMed.
- G.
M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Gottingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Wallingford, CT, 2009 Search PubMed.
- E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef.
- P. Kar, R. Haldar, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2012, 51, 4265–4273 CrossRef CAS PubMed.
- J. G. Zalatan and D. Herschlag, J. Am. Chem. Soc., 2006, 128, 1293–1303 CrossRef CAS PubMed.
- A. Husain, X. J. Yan, N. Rosales, C. Aghajanian, G. K. Schwartz and D. R. Spriggs, Clin. Cancer Res., 1997, 3, 2089–2097 CAS.
- S. Ramakrishnan, V. Rajendiran, M. Palaniandavar, V. S. Periasamy, B. S. Srinag, H. Krishnamurthy and M. A. Akbarsha, Inorg. Chem., 2009, 48, 1309–1322 CrossRef CAS PubMed.
- I. M. Ghobrial, T. E. Witzig and A. A. Adjei, Ca-Cancer J. Clin., 2005, 55, 178–194 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H spectra of compounds (Fig. S1 and S2), UV-vis spectra of compounds (Fig. S3), EPR spectrum of complex 2 (Fig. S4), mass spectra of compounds (Fig. S5–S7), π–π stacking interaction of ligand (Fig. S8), molecular packing diagram (Fig. S9), plot for rate vs. concentration of catecholase activity (Fig. S10), plot for rate vs. concentration of phosphatase activity (Fig. S11). CCDC 1418962–1418964. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra18568h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.