DOI:
10.1039/C5RA18541F
(Paper)
RSC Adv., 2015,
5, 96323-96327
A new approach to construct bulk and size-dependent continuous binary solution phase diagrams of alloys
Received
10th September 2015
, Accepted 3rd November 2015
First published on 5th November 2015
Abstract
The construction of bulk and size-dependent temperature–composition phase diagrams of alloys is critical for their industrial applications. However, the nano-phase diagrams are difficult to be determined accurately by experiments since the nano-phase equilibrium is metastable. In this work, a new approach was developed to construct both bulk and size-dependent continuous binary solution phase diagrams with three steps: (1) determining bulk atomic interaction energy by using ab initio molecular dynamics simulation; (2) calculating size-dependent melting enthalpy, melting temperature, and atomic interaction energy using a unified nanothermodynamics model; and (3) constructing phase diagrams with the above parameters, where a typical Au–Ag alloy was studied here as an example. It is found that (i) the simulated bulk atomic interaction energy is consistent with experimental data; (ii) the melting enthalpy, melting temperature, and atomic interaction energy decrease with decreasing material size for isolated nanocrystals; and (iii) the temperatures of the solidus and liquidus curves drop and the two-phase zone becomes small for the Au–Ag nanoalloy. The general approach developed here can be used to investigate other continuous binary alloy systems and can be extended to construct other phase diagrams, for example, the eutectic phase diagram.
1. Introduction
Recently nanoalloys have been receiving great attention due to their scientific and industrial importance even though single-component metallic nanocrystals have been extensively investigated. A number of reports have demonstrated that nanoalloys show superior physicochemical properties comparable to or even better than their alloy components.1–4 For example, a recent study indicated that Au–Ag alloy nanoparticles (NPs) have a higher catalytic activity than pure Au and Ag NPs for CO oxidation.1 Moreover, alloying is also an efficient approach to enhance the thermal stability of nanoscale materials.3 As a result, the construct of bulk and size-dependent phase diagrams of alloys is of fundamental importance for their fabrications and industrial applications.5–7 However, how to accurately determine the phase diagrams in experiments is a difficult task, especially at the nanometer scale, due to metastable nature of the nano-phase equilibrium.8 As a result, theoretical modeling has become an attractive alternative approach in recent years. For example, several theoretical studies have been implemented to establish solid solution phase diagrams of nanosized binary alloys, which have larger surface/volume ratio than that of bulk alloys.5,6,9–12 Wautelet et al.5 developed a model to construct phase diagrams of Ge–Si nanoparticles by considering the size-dependent melting temperature Tm(r) of pure elements, where r is the radius of nanoparticles and nanowires or half-thickness of thin films. It is found that both solidus and liquidus curves shift to lower temperatures compared to the bulk phase diagram. But, the size dependence of the melting enthalpy Hm(r) was neglected in this model. Tanaka et al.6 calculated binary phase diagrams of nanoparticles by considering the composition- and temperature-dependent excess Gibbs energies and surface tensions of the solid and liquid phases with some rough approximations. Liang et al.10 derived a model to establish phase diagrams of solid solution binary systems of metals Cu–Ni, semiconductors Ge–Si, ceramics Al2O3–Cr2O3 and V2O3–Cr2O3, and organic crystals p-chlorobromobenzene–p-dibromobenzene by considering Tm(r), Hm(r) and size-dependent atomic interaction energy Ω(r). It is found that the two-phase field shrinks with decreasing r. More recently, Guisbiers et al. presented phase diagrams of Au–Cu11 and Cu–Ni12 nanoparticles with considerations of effects of size, shape and surface segregation. Moreover, both experimental and theoretical efforts13–17 have also been implemented to understand the size-dependent eutectic phase diagrams, which are more complicated than those of the solid solution. These approaches have contributed greatly to the basic understanding of nano-phase equilibria of different alloy systems. However, to date related reports on this topic are still limited and many challenges remain even for solid solution phase diagrams. For example, the critical parameters of bulk atomic interaction energy ΩS(∞) and ΩL(∞) are not considered or obtained from the known bulk phase diagrams in the above theoretical methods,5,6,9–12 where ∞ denotes the bulk, and the superscripts L and S denote the liquid and solid, respectively. This has limited applications of these approaches.
In this work, a new approach was developed to construct both bulk and size-dependent continuous binary solution phase diagrams by combining ab initio molecular dynamics (MD) simulation and nanothermodynamics modeling. A typical Au–Ag alloy system was studied here as an example due to its widespread applications in antibacterial materials,18 catalysts,19 sensors,20 surface-enhanced Raman scattering,21,22 etc.
2. Methodology
When a binary system is in a liquid–solid equilibrium state, the chemical potentials of component A (and B) in the both phases (solid and liquid phases) are equal. As a result, the solidus and liquidus curves of Au–Ag alloy system can be expressed by:10| |
HmB(TmB − T)/TmB = ΩS(1 − xSB)2 − ΩL(1 − xLB)2 + RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(xSB/xLB) ln(xSB/xLB)
| (1a) |
| | |
HmA(TmA − T)/TmA = ΩS(xSB)2 − ΩL(xLB)2 + RTln[(1 − xSB)/(1 − xLB)]
| (1b) |
where x is the mole fraction, T absolute temperature and R ideal gas constant. Eqn (1) can be utilized to (i) determine both xLB and xSB (or xLA and xSA with xA + xB = 1) in a bulk phase diagram at a certain T when Tm, Hm, ΩS and ΩL are known; and (ii) calculate ΩS and ΩL when T, Tm, Hm, xLB and xSB are available from the bulk phase diagram. For the latter, ΩS and ΩL are generally determined at T ≈ (TmA + TmB)/2 as a first-order approximation since the composition effects on ΩS and ΩL are weak for continuous solution alloys due to small electronegativity difference between their components.10 In this case, it is evident that a well-constructed bulk phase diagram measured in experiments is necessary if one wants to establish a size-dependent phase diagram. This has impeded the development of nanothermodynamics database. In this work, ΩS(∞) and ΩL(∞) will be obtained by ab initio MD simulations.
2.1 Ab initio MD simulations
ΩS and ΩL of regular solution are given by:10| |
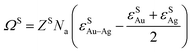 | (2a) |
| |
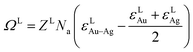 | (2b) |
where Z is the coordination number, Na the Avogadro constant, and ε the bond strength.10 ε can be determined by:| |
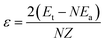 | (3a) |
| |
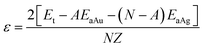 | (3b) |
where eqn (3a) and (3b) are for elemental crystals (Au and Ag) and alloys (Au50Ag50), respectively. Here, Et is the total energy, Ea the single-atom energy, N the total number of atoms, A the number of Au atoms in Au50Ag50 solid solution. Note that eqn (3) is applicable for pure metals or alloys in both solid and liquid states.
For geometry optimization, first-principles density functional theory (DFT)23,24 calculations on Au, Ag and Au50Ag50 solid solution are implemented by using CASTEP code.25 The exchange–correlation interaction was treated within the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) function.26 All 4 atoms in the unit cells are fully relaxed with a convergence tolerance of 5 × 10−6 eV per atom of energy, 0.01 eV Å−1 of maximum force, and 5 × 10−4 Å of maximum displacement. The 8 × 8 × 8 k-points27 were used to sample the Brillouin zone and the energy cutoff of 360 eV. The ultrasoft pseudopotential28 was used for all structures in the simulations. Based on the optimized systems, 3 × 3 × 3 supercells (Au, Ag and Au50Ag50 solid solution) with 108 atoms were built.
Then, the MD simulations were performed by using ab initio calculations. At first, the MD simulations were performed with NPT (dynamics with a thermostat to maintain a constant temperature and with a barostat to maintain a constant pressure). To characterize the liquid states, the constant temperatures are set as the melting points of Au, Ag and Au50Ag50, which are 1337, 1235 and 1306 K, respectively.29 The simulated lattice parameters for liquid Au, Ag and Au50Ag50 are 4.397, 4.466 and 4.436 Å, respectively. The corresponding volume changes for the melting of Au, Ag and Au50Ag50 are 25.351%, 30.575% and 28.906%, respectively, which are much larger than the reported data of 1.2–6.8% for face-centered-cubic (FCC) structures.30 Hence, these supercell structures cannot describe the liquid states accurately and the NPT method is invalid in this case. Alternatively, in this work, we conducted all MD simulations with NVT (dynamics at a fixed volume with a thermostat where a constant temperature is kept). The details will be given below.
The MD simulations with the NVT method were performed with the total simulation time and time step are 10.0 ps and 1.0 fs, respectively. The 2 × 2 × 2 k-points27 were used to sample the Brillouin zone and the energy cutoff of 300 eV. The MD simulations were performed at T = 300 K, where the lattice parameters of aS and aL for the solid and liquid states, respectively, used in the simulations are listed in Table 1. For the solid states, the simulated results of ESt and ESa, and the calculation results of εS and ΩS are also listed in Table 1. The corresponding solid structures of Au, Ag and Au50Ag50 solid solution after MD simulations are plotted in Fig. 1. For the liquid states, Fig. 2 shows the supercell structures of liquid Au, Ag and Au50Ag50 after simulations. The simulated ELt and ELa, and the calculated εL and ΩL are also given in Table 1.
Table 1 Related parameters used in the simulations and modeling
| |
Au |
Ag |
Au50Ag50 |
| εS and εL are determined by eqn (3). ΩS(∞) and ΩL(∞) are determined by eqn (2a) and (2b), respectively. For the molar weight M and the liquid density ρL of Au50Ag50, the average values of those of Au and Ag are used. Vmol = M/ρL. The atomic volume can be obtained by Va = (Vmol × 1024)/Na. The volume of unit cells V in FCC crystals is given by V = 4Va, and the lattice parameter in liquid state aL = V1/3. h = (21/2/2)a for FCC crystals. For Au and Ag, Sb = Eb/Tb. For Au50Ag50, the average value of those of Au and Ag is used. The values of Tm(r), Hm(r) and Ω(r) are calculated from eqn (4). |
| aS (Å)31 |
4.078 |
4.086 |
4.076 |
| ESt (eV) |
−98678.488 |
−110991.869 |
−104844.038 |
| ESa (eV) |
−910.646 |
−1025.035 |
|
| εS (eV)a |
−0.507 |
−0.445 |
−0.490 |
| ΩS(∞) (J mol−1)b |
|
−15828.562 |
|
| ρL (g cm−3) |
17.4 (ref. 32) |
9.15 (ref. 32) |
13.3c |
| M (g mol−1) |
196.97 (ref. 33) |
107.87 (ref. 33) |
152.42c |
| Vmol (cm3 mol−1)d |
11.320 |
11.789 |
11.460 |
| aL (Å)e |
4.221 |
4.279 |
4.239 |
| ELt (eV) |
−98684.644 |
−110989.958 |
−104845.234 |
| ELa (eV) |
−910.650 |
−1025.028 |
|
| εL (eV)a |
−0.516 |
−0.443 |
−0.492 |
| ΩL(∞) (J mol−1)b |
|
−14171.621 |
|
| h (nm)f |
0.2884 |
0.2889 |
0.2882 |
| Eb (kJ mol−1)33 |
368 |
285 |
|
| Tb (K)33 |
3129 |
2435 |
|
| Sb (J mol−1 K−1)g |
117.609 |
117.043 |
117.326 |
| Tm(r = 5 nm) (K)h |
1147.643 |
1060.509 |
|
| Hm(∞) (kJ mol−1)33 |
12.5 |
11.3 |
|
| Hm(r = 5 nm) (kJ mol−1)h |
10.730 |
9.703 |
|
| ΩS(r = 5 nm) (J mol−1)h |
|
−13592.761 |
|
| ΩL(r = 5 nm) (J mol−1)h |
|
−12169.865 |
|
 |
| | Fig. 1 Solid supercell structures of (a) Au, (b) Ag and (c) Au50Ag50 after MD simulations with NVT at 300 K. The dark and light balls represent Au and Ag atoms, respectively, and a is the lattice parameter of each structure. | |
 |
| | Fig. 2 Liquid supercell structures of (a) Au, (b) Ag and (c) Au50Ag50 after MD simulations with NVT at 300 K. | |
2.2 Nanothermodynamics modeling
In order to obtain the size-dependent phase diagrams of Au–Ag alloys, the size effects on these parameters in eqn (1), Tm, Hm and Ω, should be considered. Recently, it has been revealed that vacancy formation determined by the cohesive energy variation is one of the intrinsic factors that dominate the variation of the potential profile and thus size-dependent physicochemical properties of low-dimensional nanocrystals.8,34 Therefore, the size-dependent cohesive energy function Ec(r) dominates the size dependence of a number of physicochemical properties of nanocrystals, including Tm(r), Hm(r) and Ω(r).
Combining the Ec(r) function reported in the literature,8,34 a universal relation can be obtained for isolated nanoparticles,
| |
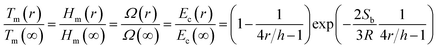 | (4) |
where
Sb =
Eb/
Tb is bulk solid–vapor transition entropy of a crystal as determined by bulk solid–vapor transition enthalpy
Eb and solid–vapor transition temperature
Tb, and
h the nearest atomic distance. Substituting
eqn (4) into
eqn (1), we can construct size-dependent Au–Ag phase diagrams.
3. Results and discussions
As shown in Table 1, the simulation result of ΩS(∞) = −15828.562 J mol−1 agrees well with the result [ΩS(∞) = −16402 J mol−1] obtained from experimental bulk Au–Ag phase diagram35 with the deviation of only 3.5%. For ΩL(∞), the corresponding values are −14171.621 and −15599 J mol−1,35 respectively, and the deviation is 9.15%. Such a deviation may arise from (i) the assumption of composition independence of Ω (only Au50Ag50 was taken into account in this work); and (ii) the density value of liquid Au50Ag50 (average value of those of Au and Ag) used in the simulations. Nonetheless, the ab initio MD simulation results of ΩS(∞) and ΩL(∞) for Au–Ag alloys exhibit relative accuracy within 10% error. These data can thus be used to establish bulk Au–Ag phase diagram later.
Fig. 3 plots (1) bulk Au–Ag phase diagrams obtained in experiments;35 (2) the calculation results of bulk and nanosized (r = 5 nm) Au–Ag phase diagrams from eqn (1), where Tm(r), Hm(r) and Ω(r) are calculated from eqn (4); and (3) experimental data of Tm(r) of Au nanoparticles (r = 5 nm)36 denoted as the symbols ∇ [Tm(r) = 1169.4 ± 63.5 and 1180.4 ± 88.2 K] and a MD simulation result of Tm(r) of Ag nanoparticles (r = 5 nm)37 denoted as the symbol Δ [Tm(r) = 1112.1 K]. As shown in the figure, the calculated bulk Au–Ag phase diagram from eqn (1), where the simulation results of ΩS(∞) and ΩL(∞) are used in the modeling, is consistent with that measured in experiments within 3% error. Thus, our method provides a simple and effective way to calculate the interaction energy of continuous binary alloys.
 |
| | Fig. 3 Bulk and nanosized (r = 5 nm) Au–Ag phase diagrams. The solid lines denote predictions from eqn (1). The symbols ● and ○ are experimental data of the solidus and liquidus curves, respectively.35 The symbols ∇ are experimental data of Tm(r) for Au nanoparticles (r = 5 nm)36 and the symbol Δ is a MD simulation result of Tm(r) for Ag nanoparticles (r = 5 nm).37 | |
It is clear that Tm(r), Hm(r) and Ω(r) all decrease with decreasing r according to eqn (4), indicating the instability of isolated nanoparticles compared with their counterparts in bulk materials. From Fig. 3, we can see that the model predictions of Tm(r) are consistent with the experimental data of Au nanoparticles (r = 5 nm). Moreover, the derivation is only 5% between our calculation and the MD simulation results of Tm(r) for Ag nanoparticles (r = 5 nm). These demonstrate the accuracy of eqn (4). As r reducing, the surface/volume ratio increases, resulting in the formation of a large number of surface dangling bonds. Thus, the surface atoms are in a higher energetic state than those of the interior atoms, depressing Ec(r), Tm(r), Hm(r) and Ω(r).
As shown in Fig. 3, the decreased Tm(r) results in the drop of the solidus and liquidus curves, and the reduced Ω(r) causes the shrinkage of the two-phase zone in nanosized Au–Ag phase diagram compared with that of the bulk. The constructed size-dependent phase diagrams and these findings would be validated in future experimental studies. Our calculation results are critical for the basic understanding of the phase transition theory of nanoalloys and also for their scale-up industrial applications. If the necessary parameters are available, the general approach developed in this work could be used to establish other continuous binary alloy systems, and also be extended to construct other types of phase diagrams, for instance, the eutectic phase diagram. Moreover, it should be noted that only the size effect was focused in this work for a simplification. The dimensionality and shape effects could also be considered in our nanothermodynamics models,8,34 which will be explored in future studies.
4. Conclusions
In summary, a simple and effective approach was developed to establish both bulk and size-dependent Au–Ag phase diagrams based on (1) bulk interaction energy Ω(∞) calculated by ab initio MD simulations; and (2) size-dependent melting temperature Tm(r), melting enthalpy Hm(r), and interaction energy Ω(r) using a unified nanothermodynamics model. It is found that the simulated Ω(∞) values are consistent with the results obtained from experimental bulk Au–Ag phase diagram. Tm(r), Hm(r) and Ω(r) all decrease with decreasing r for isolated Au and Ag nanoparticles, resulting in the drop of solidus and liquidus curves and shrinkage of the two-phase zone in nanosized Au–Ag phase diagram. The developed general approach could also be used to construct different phase diagrams in other alloy systems if the relevant parameters are available.
Acknowledgements
We wish to thank ChangBai Mountain Scholars Program and Jilin University Basic Research Grants Program for financially supporting this project and the computing resources of High Performance Computing Center of Jilin University and National Supercomputing Center in Jinan, China. We also thank G. Guisbiers for helpful discussions.
References
- C. W. Yen, M. L. Lin, A. Q. Wang, S. A. Chen, J. M. Chen and C. Y. Mou, J. Phys. Chem. C, 2009, 113, 17831–17839 CAS.
- M. Oezaslan, M. Heggen and P. Sreasser, J. Am. Chem. Soc., 2012, 134, 514–524 CrossRef CAS PubMed.
- T. Chookajorn, H. A. Murdoch and C. A. Schuh, Science, 2012, 337, 951–954 CrossRef CAS PubMed.
- C. H. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- M. Wautelet, J. P. Dauchot and M. Hecq, Nanotechnology, 2000, 11, 6–9 CrossRef CAS.
- T. Tanaka and S. Hara, Z. Metallkd., 2001, 92, 1236–1241 CAS.
- A. van de Walle, Q. Hong, S. Kadkhodaei and R. Sun, Nat. Commun., 2015, 6, 7559 CrossRef CAS PubMed.
- Q. Jiang and Z. Wen, Thermodynamics of Materials, Higher Education Press, Springer-Verlag, Beijing, Berlin Heidelberg, 2011 Search PubMed.
- R. Vallee, M. Wautelet, J. P. Dauchot and M. Hecq, Nanotechnology, 2001, 12, 68–74 CrossRef CAS.
- L. H. Liang, D. Liu and Q. Jiang, Nanotechnology, 2003, 14, 438–442 CrossRef CAS.
- G. Guisbiers, S. M. Rosales, S. Khanal, F. R. Zepeda, R. L. Whetten and M. J. Yacaman, Nano Lett., 2014, 14, 6718–6726 CrossRef CAS PubMed.
- G. Guisbiers, S. Khanal, F. R. Zepeda, J. R. de la Puente and M. J. Yacaman, Nanoscale, 2014, 6, 14630–14635 RSC.
- W. A. Jesser, R. A. Shneck and W. W. Gile, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 144121 CrossRef.
- J. G. Lee and H. Mori, Phys. Rev. Lett., 2004, 93, 235501 CrossRef CAS PubMed.
- J. Weissmuller, P. Bunzel and G. Wilde, Scr. Mater., 2004, 51, 813–818 CrossRef.
- T. Ivas, A. N. Grundy, E. P. Karadeniz and L. J. Gauckler, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2012, 36, 57–64 CrossRef CAS.
- H. M. Lu and X. K. Meng, Sci. Rep., 2015, 5, 11263 CrossRef PubMed.
- M. Banerjee, S. Sharma, A. Chattopadhyay and S. S. Ghosh, Nanoscale, 2011, 3, 5120–5125 RSC.
- H. L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS PubMed.
- J. I. Francisco and P. Z. Francis, ACS Nano, 2008, 2, 1543–1552 CrossRef PubMed.
- M. K. Fan, F. J. Lai, H. L. Chou, W. T. Lu, B. J. Hwang and A. G. Brolo, Chem. Sci., 2013, 4, 509–515 RSC.
- A. H. Noor, R. H. Benjamin and A. F. David, Chem. Commun., 2014, 50, 12389–12391 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- J. P. Perdew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1983, 4, 30–38 CrossRef.
- H. M. Lu and Q. Jiang, Phys. Status Solidi B, 2004, 241, 2472–2476 CrossRef CAS.
- V. A. Lubarda, Mech. Mater., 2003, 35, 53–68 CrossRef.
- J. Brillo, I. Egry and I. Ho, Int. J. Thermophys., 2006, 27, 494–506 CrossRef CAS.
- http://www.webelements.com/ (Web Elements Periodic Table).
- C. C. Yang and Y.-W. Mai, Mater. Sci. Eng., R, 2014, 79, 1–40 CrossRef.
- S. Hassam, M. Gambino, E. Gaune, J. P. Bros and J. Agren, Metall. Trans. A, 1988, 19, 409–416 CrossRef.
- K. Dick, T. Dhanasekaran, Z. Zhang and D. Meisel, J. Am. Chem. Soc., 2002, 124, 2312–2317 CrossRef CAS PubMed.
- W. Luo, W. Hu and S. Xiao, J. Phys. Chem. C, 2008, 112, 2359–2369 CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 







