
Synthesis of chitosan-coated polyoxometalate nanoparticles against cancer and its metastasis
Abstract
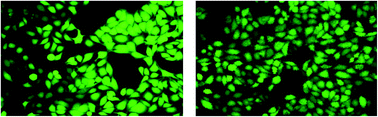
Three different Keggin-type polyoxometalates (POMs) [PW12O40]3−, [TiW11CoO40]7−, and [Ti2PW10O40]7−, were synthesized and then encapsulated in chitosan to prepare nanoparticles, CS–PW12, CS–TiW11Co, and CS–Ti2PW10. The synthesized nanoparticles were physicochemically characterized in terms of particle size, zeta potential, entrapment efficiency and in vitro release of the entrapped POM. The most efficient formulation was CS–TiW11Co, with a particle size of 105 ± 6 nm and an entrapment efficiency of 87 ± 12 (%). The CS–TiW11Co nanoparticles showed the highest activity when tested against tissue nonspecific alkaline phosphatase (TNAP) with IC50 = 102.0 ± 9.68 nM. The anticancer potential of the free POMs and their nanoparticles were also studied and CS–TiW11Co showed the highest inhibition (IC50 = 1.06 ± 0.09) on HeLa cells. To observe signs of apoptosis in HeLA cells, DAPI staining was performed after treatment with CS–TiW11Co nanoparticles. Furthermore, the reactive oxygen species (ROS) production was examined by H2DCF-DA dye under a fluorescence microscope. Our study revealed that CS–TiW11Co nanoparticles are very effective in cancer treatment and its associated metastasis especially in osteoblastic lesions with minimal adverse effects on normal cells (Vero cells).
 Please wait while we load your content...
Please wait while we load your content...

