
A quick and easy synthesis of fluorescent iron oxide nanoparticles featuring a luminescent carbonaceous coating via in situ pyrolysis of organosilane ligands†
M. Worden‡
a,
L. Bergquistb and
T. Hegmann*ab
aDepartment of Chemistry and Biochemistry, Kent State University, Kent, OH, 44240-0001 USA
bChemical Physics Interdisciplinary Program, Liquid Crystal Institute, Kent State University, Kent, OH, 44240-0001 USA. E-mail: thegmann@kent.edu
First published on 17th November 2015
Abstract
We report a simple, two-step method for making magnetic, photoluminescent iron oxide (magnetite) core/carbonaceous shell nanoparticles emitting blue light. The core–shell nanoparticles are created by an aqueous synthesis and functionalized in situ with three different organosilanes. Pyrolysis of the organosilane ligand shell at 200 °C yields water-dispersible core–shell particles visibly fluorescing under UV light.
Introduction
Combinations of carbonaceous nanoparticles (NPs) or carbon dots (Cdots) with metal and metal oxide NPs receive increasing attention, particularly when the beneficial properties of both constituents are required (e.g., magnetic and lumincescent). Typically this involves functionalizing or doping the Cdot core with metal or metal oxide nanostructures, rather than vice versa. Examples include combinations of Cdots with ZnO and ZnS,1 as well as with Au, Cu, and Pd.2 Li et al. synthesized titania and silica NPs with Cdots physisorbed onto their surface for photocatalytic applications.3 The same group also reported a similar strategy for creating magnetic iron oxide NPs (IONPs) coated with Cdots.4 In each of these cases the core particles are several hundred nanometers in size, and the metal oxide NPs and Cdots must be made separately before being combined in a composite material. Wang et al. created smaller clusters of IONPs of approximately 50 nm in size decorated with Cdots through a one-pot solvothermal method.5 The synthesis involved a reaction between ferrocene and concentrated hydrogen peroxide in an autoclave, and required 48 hours at 200 °C.Here we report a straightforward method for making magnetic IONPs featuring a carbonaceous shell that furnishes blue-light emitting fluorescence properties. The IONPs are created by a simple aqueous synthesis reported by our group earlier6 and are functionalized in situ with hydrophilic silane compounds.7 Pyrolysis of the attached siloxane shell at 200 °C yields water-dispersible particles that visibly fluoresce under UV light.
Cdots (referred to as carbogenic NPs) are a relatively new type of fluorescent nanomaterial. Unlike the more well-known and ubiquitous quantum dots (QDs) that exhibit size-dependent photoluminescence, Cdots show excitation wavelength-dependent photoluminescence in the visible spectrum. In contrast to QDs, which typically contain heavy metals, Cdots are comprised solely of organic molecules. This fact has led to investigations into Cdots for bioapplications, such as cell imaging and labelling, from which quantum dots are often restricted.8
Early syntheses of these materials involved non-chemical modification of carbon precursors, such as laser ablation of graphite or through electrochemical decomposition of carbon nanotubes.9 Subsequent investigations have allowed for the synthesis of Cdots through solely chemical means. Bourlinos et al. synthesized Cdots through a simple pyrolysis of various ammonium salts.10 A typical synthesis involved creating a salt of citric acid and a long chain amine, followed by high temperature pyrolysis of the salt at several hundred degrees for a few hours. The Cdots were highly monodisperse, and could be made hydrophilic or hydrophobic, depending on the exact composition of the precursors. Wang et al. reported similar methods for Cdot synthesis in which they decomposed citric acid with molten lithium nitrate under argon11 as well as a high temperature thermolysis of citric acid in a mixture of octadecene and hexadecylamine.12 The same group as well as another group also recently reported a synthesis of Cdots in which the amine group was attached to an organosilane compound (often termed Si-Cdots).13 They proposed that the Si-Cdots from this synthesis are composed of a mixed amide core, resulting from the acylation of the amine group, with the trimethoxysilane group remaining as a functionalizing group on the surface of the NPs.
This particular work inspired the idea that it may be possible to create core–shell IONP with a photoluminescent carbogenic or carbonaceous shell materials through the simple pyrolysis of silanized IONPs, described herein.
The exact mechanism behind the photoluminescent properties of Cdots is still under debate. One possible mechanism is the formation of graphene-like sheets within the particle, which would thus mimic the PL process as seen in reduced graphene and graphene oxide (GO). In this process, the fluorescence arises not from quantum confinement effects, as in QDs, but rather from interactions between localized clusters, or “islands”, of sp2 hybridized carbons.14 When these clusters are in close proximity – but not touching, so as to avoid quenching – such as in stacks of graphene sheets, the band gap between π and π* orbitals is lowered enough so that electrons can be excited by UV radiation. Additionally, recent research on graphene oxide has revealed that C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups play an important role in tuning the band gap energy and the overall emission wavelengths seen in GO.15 Other researchers have noted that passivation by a variety of chemical functionalities can help increase the PL intensity, as well as contribute to emission wavelength tunability, in Cdots.16 This is often attributed to a second possible fluorescence mechanism involving surface defects on the particles. Defects on the surface of Cdots create carbon clusters with different hybridizations, resulting in PL emission properties similar to that seen in graphene and GO without the explicit formation of these chemical structures.17 A recent investigation by Gan et al. attempted to elucidate the PL mechanism in blue-light emitting Cdots.18 They found that the surface defect mechanism dominates for Cdots that emit predominately blue light, while Cdots showing more tunability in emission wavelengths have fewer defect sites. Thus the PL mechanism is very dependent on the specific nature of the Cdots in terms of size, internal composition, and surface functionalities, and can even depend on the synthetic method used.19
O functional groups play an important role in tuning the band gap energy and the overall emission wavelengths seen in GO.15 Other researchers have noted that passivation by a variety of chemical functionalities can help increase the PL intensity, as well as contribute to emission wavelength tunability, in Cdots.16 This is often attributed to a second possible fluorescence mechanism involving surface defects on the particles. Defects on the surface of Cdots create carbon clusters with different hybridizations, resulting in PL emission properties similar to that seen in graphene and GO without the explicit formation of these chemical structures.17 A recent investigation by Gan et al. attempted to elucidate the PL mechanism in blue-light emitting Cdots.18 They found that the surface defect mechanism dominates for Cdots that emit predominately blue light, while Cdots showing more tunability in emission wavelengths have fewer defect sites. Thus the PL mechanism is very dependent on the specific nature of the Cdots in terms of size, internal composition, and surface functionalities, and can even depend on the synthetic method used.19
Results and discussion
A schematic representation of the overall reaction sequence is depicted in Fig. 1. The accompanying pictures of the mixed ligand shell 3-aminopropyltriethoxysilane (AMS) and N-(tri-methoxysilylpropyl)ethylenediaminetriacetate (EDTS) coated IONPs after purification and pyrolysis show that the resulting core–shell NPs can be redispersed in water (when sonicated) and that the solution is visibly photoluminescent when illuminated under 365 nm UV light. Dispersions of the precursor IO NPs before pyrolysis do not show any measurable photoluminescence, indicating that the pyrolysis process, performed at a temperature below any decomposition of the ligand shell is recorded by thermogravimetric analysis (TGA), is integral in the modification of the IONP surface. The final luminescent IONPs are attracted to rare earth magnets (used to isolate and purify the NPs) and can easily be redispersed by shaking or mild sonication resulting in stable suspensions with bright blue photoluminescent IO core/carbonaceous shell NPs.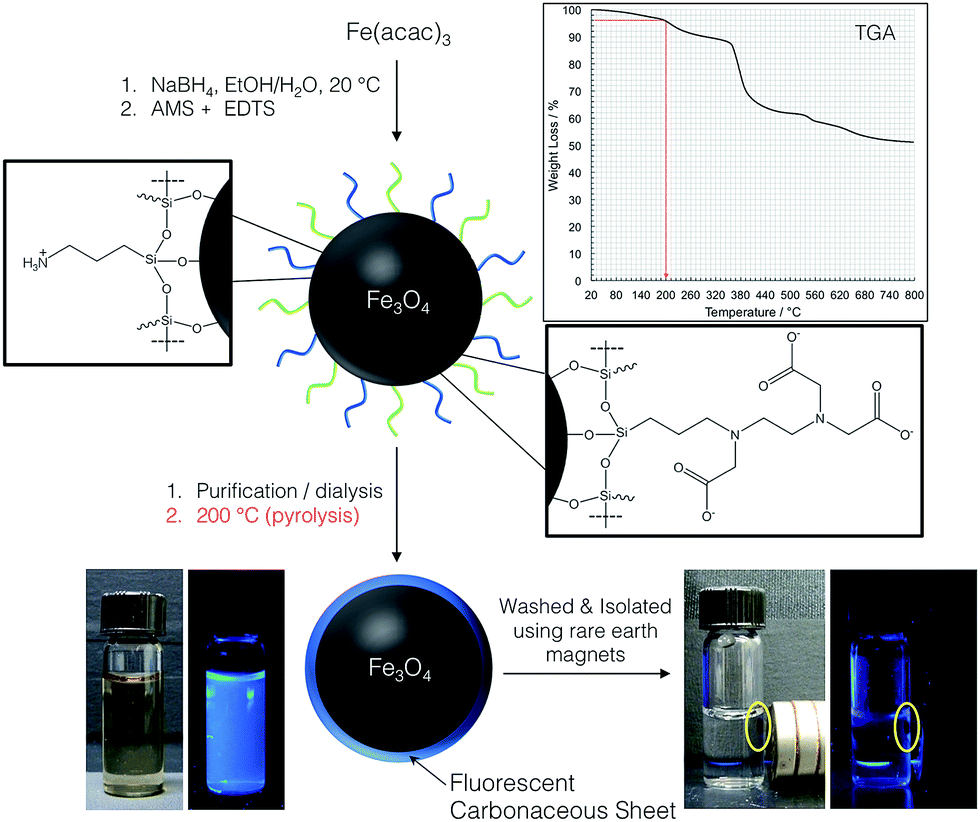 | ||
| Fig. 1 Schematic showing the process for creating IONP with a carbonaceous shell in two simple steps. Pyrolysis of the ligand shell of the mixed monolayer-capped IO-NPs at 200 °C results in IONPs with carbonaceous shell that luminesce when illuminated under 365 nm UV light. The final core–shell IONPs are attracted to a rare earth magnet, and the luminescence of the solution vanishes. In such close contact, the luminescence of the IONPs with carbonaceous shell is quenched (bottom right), but reoccurs as soon as the particles are redispersed in solution by shaking the vial or mild sonication. | ||
Essentially the same result was achieved for IONPs capped exclusively with either one of the silanes (AMS or EDTS), with the only detectable difference being the photoluminescence intensity of the final core/shell IONPs. The photoluminescent properties of each core/shell IONP set dispersed in water, before and after pyrolysis, were measured by fluorescence spectroscopy. The particle dispersions were excited with light at wavelengths between 250 and 400 nm, in 25 nm increments. Fig. 2 shows the results for the initially mixed monolayer-capped IONPs (AmS-EDTS-IONPs); Fig. 3 and 4 show the results for the other two silane-capped IONPs (AMS-IONP and EDTS-IONPs), respectively.
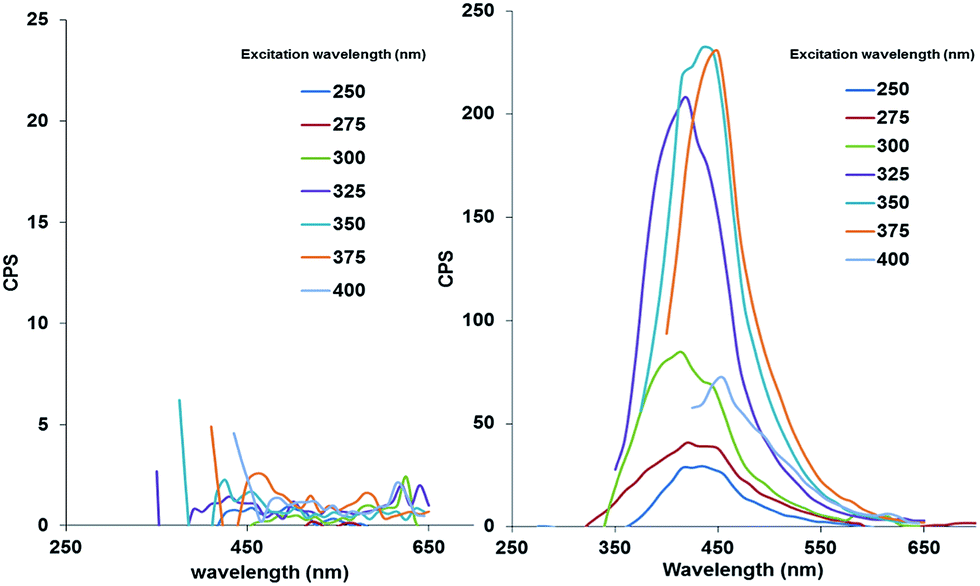 | ||
| Fig. 2 Fluorescence spectra for AmS-EDTS-IONPs before (left; showing practically only noise) and after pyrolysis (right). | ||
 | ||
| Fig. 3 Fluorescence spectra for AmS-IONPs before (left; showing practically only noise) and after pyrolysis (right). | ||
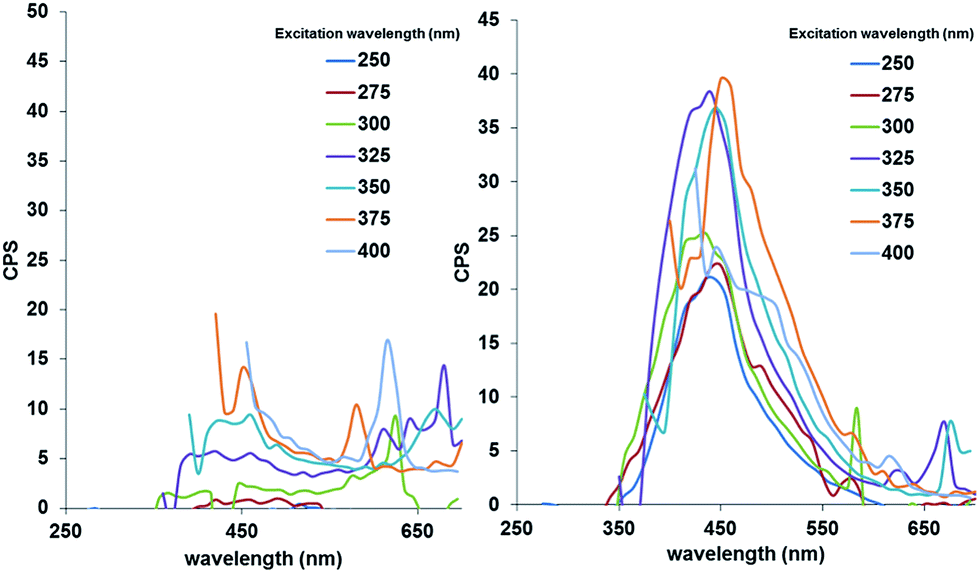 | ||
| Fig. 4 Fluorescence spectra for EDTS-IONPs before (left; showing practically only noise) and after pyrolysis (right). | ||
In each case, the initial NP dispersions before pyrolysis show no detectable photoluminescence. Post-pyrolysis, each particle set displays significantly enhanced photoluminescence, with a slight difference in excitation wavelength corresponding to the maximum emission peak. The AmS-EDTS-IONPs after pyrolysis show narrower, more symmetrical peaks with maximum emission peaks centered around 400 nm corresponding to 325, 350, and 375 nm excitation wavelengths (Fig. 2). The AmS-IONPs after pyrolysis display broader, more asymmetrical emission peaks between 400 and 450 nm, with a maximum emission related to a 325 nm excitation wavelength (Fig. 3). Finally, EDTS-IONPs after pyrolysis show the least intense emission peaks of the three, with broad, asymmetrical emission peaks centered around 450 nm, with a maximum emission roughly equal between 325, 350 and 375 nm (Fig. 4).
TEM and HR-TEM imaging was performed on each set of IONPs before and after pyrolysis. No visible differences in particle size or morphology were detectable for any set of particles (Fig. 5). This is not necessarily surprising, however, since the IO core, only approximately 5 nm in diameter, provides the majority of electron density and thus image contrast under TEM. The thin organosilane surface ligands before pyrolysis do not provide sufficient contrast to be seen. The carbonaceous coatings after pyrolysis, however, can be detected by HR-TEM imaging as shown in Fig. 6. In these TEM images, the crystalline nature of the IONP core is clearly visible. More importantly, however, the amorphous nature of the thin (0.5 to 0.8 nm on average), carbonaceous Si–C shell around many of the IONPs is also clearly discernable (particularly of those that are in clear focus, yellow arrows in Fig. 6). Overall, the TEM images do demonstrate that the IONPs have not been destroyed by the pyrolysis, and that the particles remain individually separated.
 | ||
| Fig. 5 Representative TEM images of silanized AMS-EDTS-IONPs: (a) before and (b) after pyrolysis. The average size of these IONPs is 5 ± 0.8 nm. | ||
 | ||
| Fig. 6 HR-TEM images of the previously silanized IONPs after pyrolysis: (a) former AMS-IONPs, (b) former EDTS-IONPs, and (c) former AMS-EDTS-IONPs. The thickness of the Si–C carbonaceous shell can be estimated to about 0.5 to 0.8 nm. For additional HR-TEM images, see ESI.† | ||
We also performed energy-dispersive X-ray spectroscopy (EDS, or EDX), which confirmed the presence of C, O, Si and Fe as expected (see ESI†). The non-ideal nature of these NP samples (rough, inhomogeneous, amorphous), though, inhibited a true quantitative analysis via EDS as highlighted in a recent review in the technique by Newbury and Ritchie.20
To obtain quantitative data, we performed elemental analysis (EA) on all IONPs before and after pyrolysis (see ESI†). These data show that elements such as H and N, which are able to leave the sample in gaseous H2 and N2 form during pyrolysis, are less abundant after pyrolysis for the two IONPs capped with the AMS silane prior pyrolysis. For the EDTS-IONPs, the concentration of H and N remained almost constant. The abundance of C increased for the AMS-IONPs and significantly decreased for the mixed silane-shell AMS-EDTS-IONPs after pyrolysis. In the case of the EDTS-IONPs, the values for H and N were practically identical before and after pyrolysis, and the value for C increased slightly. Considering this data, we can perhaps conclude that the mixed silane AMS-EDTS-IONPs undergo decarboxylation when these IONPs are heated to 200 °C. A similar decline of C in the elemental composition was not observed for the EDTS-IONPs, and seems to indicate that these IONPs with densely packed EDTS silanes at the IONP surface require higher pyrolysis temperatures or longer pyrolysis times to form a more pronounced carbonaceous shell. This is also supported by the much reduced luminescence intensity measured for these EDTS-IONPs shown in Fig. 4. However, the two IONPs capped with AMS, similar to reports using the methoxy equivalent of AMS (3-aminopropyltrimethoxysilane, APTMS) to produce bright luminescent Cdots, are capable of forming a more intense photoluminescent carbonaceous shell around the IONP core.
Combined, HR-TEM, EDS and EA data nevertheless support the formation of a Si–C carbonaceous shell around the IONPs that can likely be further optimized for higher photoluminescence intensities and composition by longer pyrolysis times or higher temperatures.
Finally, FT-IR spectroscopy was used to characterize the surface chemistry of the particles before and after pyrolysis (Fig. 7). Surprisingly, the pyrolysis process causes little notable differences in surface functionality of the particles. In Fig. 7a, AmS-IONPs before and after pyrolysis of the capping silane coating are compared. The only observable differences between the spectra involve the relative intensity of the broad peaks between 3200 and 3500 cm−1, but this is likely due to changes in the amount of adsorbed moisture, which causes the O–H vibrational stretch. Fig. 7b shows spectra for the AmS-EDTS-IONPs before and after pyrolysis. Once again the same functional groups are present in each spectrum. The only significant difference involves a decrease in relative intensity of the broad peak centered at approximately 1610 cm−1 after pyrolysis. This peak can be associated with both asymmetric vibrational stretching of carboxylate groups (decrease in intensity supporting partial decarboxylation during pyrolysis) and scissoring of protonated amine groups, which would both largely overlap any contribution from non-amorphous –CC– sp2 clusters of the formed Si–C carbonaceous shell,21 whose formation is unlikely considering the short pyrolysis time of 45 minutes.22 Fig. 7c shows spectra for the EDTS-IONPs, again before and after pyrolysis. As before, there is little evidence of significant changes in surface functionality between the three particle sets.
 | ||
| Fig. 7 FT-IR spectra of: (a) EDTS-AMS-IONPs, (b) AMS-IONPs, and (c) EDTS-IONPs before (bottom spectrum) and after pyrolysis (top spectrum). | ||
In their synthesis of organosilane-based Cdots, Wang et al. note, through FT-IR, the formation of amide groups as the main significant difference in chemical moieties between the precursors and the Cdot products.13 There is no clear evidence for that in the IO core/carbonaceous shell NPs here, although it is possible that such a peak – centered at 1654 cm−1 – could be masked by peaks associated with asymmetric vibrational stretching of carboxylate groups and scissoring of protonated amine groups.
Conclusions
While the results herein make it difficult to determine with complete certainty the precise mechanism behind the PL properties of the IO core/carbonaceous shell NPs, some broad conclusions about the nature of these materials can be made. From TEM, there is no evidence of the formation of separate or isolated Si-Cdots on the surface of the IONPs, or of any signs that the particles have become clustered due to reactions perhaps between the surfaces of adjacent nanoparticles. This suggests that any reactions or decomposition processes arising from the pyrolysis process occur on the surface of individual IONPs. Additionally, the PL data show that the emission wavelengths are largely not varying with respect to changes in the excitation wavelength. Rather, the composite or core–shell particles emit predominately blue light in a much more narrow wavelength range than typically observed for Cdots.Differences in surface composition affect the PL emission spectra more than does the excitation wavelength. With these points in mind, quantum confinement effects arising from changes in Si-Cdot or doping level size can be eliminated.
Interactions between graphene sheets would also be expected to cause some degree of emission wavelength tunability seen in other composites containing such material, and so this mechanism can also be eliminated.
As such, we suggest that the formation of highly amorphous sp2 defect carbon clusters in thin layers on the surface of the IONPs is the primary contributor to the PL properties observed for these IO core/carbonaceous shell NPs. Further work needs to be done, however, to elucidate the precise effect that particular functional groups have on this process, as well as the effect that additional passivation with functional molecules post-pyrolysis may have on PL intensity and their overall usefulness as multi-potent medical tracers. The synthesis overall is straightforward and simple, leading to luminescent core–shell IONPs in just two simple steps without the need for special surface chemistry needed to attach a separate chromophore to create traceable NP for (bio-)medical and pharmacological applications using a combination of magnetic particle imaging (MPI), magnetic resonance imaging (MRI), and fluorescence confocal imaging.
Experimental details
Materials and methods
Iron(III) acetylacetonate (99%) was purchased from Fluka, sodium borohydride (98%) and (3-aminopropyl)triethoxysilane (AMS, 99%) from Sigma-Aldrich. N-(Trimethoxysilylpropyl)ethylenediaminetriacetate trisodium salt (EDTS) was obtained from Gelest, Morrisville, PA, USA. Millipore deionized (DI) water (R = 18 MΩ) was used in all reaction and purification steps. Transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS) was done with a FEI Tecnai TF20 TEM instrument at an accelerating voltage of 200 kV. High-resolution TEM imaging was performed using an FEI Tecnai F30 TEM instrument at an accelerating voltage of 300 kV. Particle samples were dispersed in methanol and dropcast onto 400-mesh carbon coated copper grids and air-dried prior to analysis. Fourier-transform infrared spectroscopy (FT-IR) was performed using a Nicolet Magna-IR Spectrometer 550 (Thermo Fisher Scientific, Waltham, MA, USA). Sample preparation involved pelletizing the dry silanized IONPs before and after pyrolysis in KBr. Thermogravimetric analysis was done with a TA Instruments (New Castle, DE, USA) TGA Q500. The heating rate was set at 10 °C min−1. Powdered samples were typically dried in a vacuum oven at 50 °C for 2 hours before analysis in order to eliminate any surface water. However, some initial weight loss detectable in the TGA plots is a result of additional water desorption, after which some molecular segments of both siloxane coating begin to burn off at a temperature of about 200 °C, which is why we performed the pyrolysis at this temperature. Photoluminescence (fluorescence) spectra were collected using a Varian Cary 205 Eclipse with variable excitation wavelengths. A TruSpec®Micro elemental series instrument (LECO Corp.) was used for elemental analysis of the IONPs before and after pyrolysis.Silanized IONPs
A typical experiment was conducted as follows. IONPs were first synthesized following the reduction-hydrolysis method by Yathindranath et al.6 These bare particles were then functionalized with some combination of the two silane compounds: (3-aminopropyl)triethoxysilane (AMS) and N-(tri-methoxysilylpropyl)ethylenediamine-triacetate (EDTS), following the method published by Yathindranath et al.7 This was done by injecting the silane solution directly into the reaction medium containing the IONPs 1 hour after IO (Fe3O4 – magnetite6) NP formation. Three sets of particles were made this way. The first IONP used AMS (AmS-IONPs), the second both AMS and EDTS in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (AmS-EDTS-IONPs), and the third solely EDTS (EDTS-IONPs). In each case the concentrations of silane to iron precursor was kept at a 10:1 molar ratio during the reaction. The particles in each case were purified by dialysis against water, and then isolated by evaporating the water under reduced pressure. The dried powders were then washed several times with water/ethanol mixtures and collected with a rare earth magnet before being dried in a vacuum oven. Portions of each of the purified particles were kept as a powder to be used in the subsequent pyrolysis step.
:1 molar ratio (AmS-EDTS-IONPs), and the third solely EDTS (EDTS-IONPs). In each case the concentrations of silane to iron precursor was kept at a 10:1 molar ratio during the reaction. The particles in each case were purified by dialysis against water, and then isolated by evaporating the water under reduced pressure. The dried powders were then washed several times with water/ethanol mixtures and collected with a rare earth magnet before being dried in a vacuum oven. Portions of each of the purified particles were kept as a powder to be used in the subsequent pyrolysis step.
Pyrolysis
Samples of the dried powders obtained above were placed in an oven under air at 200 °C for at least 45 minutes. The resulting black powders, washed and isolated using rare earth magnets, can be re-dispersed in water using an ultrasonication horn (at a concentration of about 1 mg mL−1) for fluorescence spectroscopy, or used as dry powders for TEM imaging and FT-IR spectroscopy.Acknowledgements
This work was financially supported by the Ohio Third Frontier (OTF) program for Ohio Research Scholars “Research Cluster on Surfaces in Advanced Materials” (support for T. H.), which also supports the cryo-TEM facility at the Liquid Crystal Institute (KSU), where some of the TEM data were acquired. We would also like to acknowledge access to the HR-TEM facility at the Swagelok Center for Surface Analysis of Materials at Case Western Reserve University, and thank Danqi Wang for expert support with image acquisition.References
- Y. Sun, X. Wang, F. Lu, L. Cao, M. J. Meziani, P. G. Luo, L. Gu and L. M. Veca, J. Phys. Chem. C, 2008, 112, 18295–18298 CrossRef CAS PubMed.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. Tsang, X. Yang and S. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- H. Zhang, H. Ming, S. Lian, H. Huang, H. Li, L. Zhang, Y. Liu, Z. Kang and S. Lee, Dalton Trans., 2011, 40, 10822–10825 RSC.
- H. Wang, Z. Wei, H. Matsui and S. Zhou, J. Mater. Chem. A, 2014, 2, 15740–15745 RSC.
- V. Yathindranath, L. Rebbough, D. F. Moore, D. W. Miller, J. van Lierop and T. Hegmann, Adv. Funct. Mater., 2011, 21, 1457–1464 CrossRef CAS.
- (a) V. Yathindranath, Z. Sun, M. Worden, L. J. Donald, J. A. Thliveris, D. W. Miller and T. Hegmann, Langmuir, 2013, 29, 10850–10858 CrossRef CAS PubMed; (b) Z. Sun, V. Yathindranath, M. Worden, J. A. Thliveris, S. Chu, F. E. Parkinson, T. Hegmann and D. W. Miller, Int. J. Nanomed., 2013, 8, 961–970 CrossRef PubMed; (c) Z. Sun, M. Worden, Y. Wroczynskyj, V. Yathindranath, J. van Lierop, T. Hegmann and D. W. Miller, Int. J. Nanomed., 2014, 9, 3013–3026 CrossRef PubMed; (d) M. Worden, M. A. Bruckman, M.-H. Kim, N. F. Steinmetz, J. M. Kikkawa, C. LaSpina and T. Hegmann, J. Mater. Chem. B, 2015, 3, 6877–6884 RSC.
- (a) S. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef CAS PubMed; (b) S. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J. Liu, Y. Liu, M. Chen, Y. Huang and Y. Sun, J. Phys. Chem. C, 2009, 113, 18110–18114 CrossRef CAS PubMed.
- (a) Y. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed; (b) J. Zhou, C. Booker, R. Li, X. Zhou, T. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef CAS PubMed.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455–458 CrossRef CAS PubMed.
- F. Wang, M. Kreiter, B. He, S. Pang and C. Liu, Chem. Commun., 2010, 46, 3309–3311 RSC.
- F. Wang, S. Pang, L. Wang, Q. Li, M. Kreiter and C. Liu, Chem. Mater., 2010, 22, 4528–4530 CrossRef CAS.
- (a) F. Wang, Z. Xie, H. Zhang, C. Liu and Y. Zhang, Adv. Funct. Mater., 2011, 21, 1027–1031 CrossRef CAS; (b) P.-C. Chen, Y.-N. Chen, P.-C. Hsu, C.-C. Shih and H.-T. Chang, Chem. Commun., 2013, 49, 1639–1641 RSC; (c) C.-C. Shih, P.-C. Chen, G.-L. Lin, C.-W. Wang and H.-T. Chang, ACS Nano, 2015, 9, 312–319 CrossRef CAS PubMed.
- G. Eda, Y. Lin, C. Mattevi, H. Yamaguchi, H. Chen, I. Chen, C. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- J. Shang, L. Ma, J. Li, W. Ai, T. Yu and G. G. Gurzadyan, Sci. Rep., 2012, 2, 792 CrossRef PubMed.
- (a) S. Chandra, S. H. Pathan, S. Mitra, B. H. Modha, A. Goswami and P. Pramanik, RSC Adv., 2012, 2, 3602–3606 RSC; (b) Y. Yang, J. Cui, M. Zheng, C. Hu, S. Tan, Y. Xiao, Q. Yang and Y. Liu, Chem. Commun., 2012, 48, 380–382 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- Z. Gan, X. Wu and Y. Hao, CrystEngComm, 2014, 16, 4981–4986 RSC.
- S. Baker and G. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- D. E. Newbury and N. W. M. Ritchie, Scanning, 2013, 35, 141–168 CrossRef CAS PubMed.
- R. de Palma, S. Peeters, M. J. van Bael, D. R. Van, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- Z. Gun, X. Wu and Y. Hao, CrystEngComm, 2014, 16, 4981–4986 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18382k |
| ‡ Current address: Department of Chemistry, Boston University, Boston, MA (USA). |
| This journal is © The Royal Society of Chemistry 2015 |