
Band structure engineering of multiple band degeneracy for enhanced thermoelectric power factors in MTe and MSe (M = Pb, Sn, Ge)
Guangqian Ding,
Jie Li and
Guoying Gao*
School of Physics and Wuhan National High Magnetic Field Center, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: guoying_gao@mail.hust.edu.cn
First published on 22nd October 2015
Abstract
The thermoelectric (TE) conversion efficiency is always limited by a low TE figure of merit (ZT). Improving ZT requires both a high power factor (PF) and a low thermal conductivity. So far, however, most efforts to improve ZT have been made by reducing the thermal conductivity rather than maximizing the PF. Recently, band engineering which can effectively solve the paradox between the density-of-states effective mass and carrier mobility, has been treated as an efficient approach to improve ZT by maximizing the PF. In this paper, based on first-principles and the Boltzmann transport theory, we calculate the electronic structure and thermoelectric properties of classical IV–VI semiconductors MTe and MSe (M = Pb, Sn, Ge). We find that band engineering of multiple band degeneracy induced by engineering the conduction bands near the Fermi level can increase the room temperature n-type PF around 3 to 8 times. The present work is useful in thermoelectrics and will attract more research interest in optimizing the TE performance by band engineering.
1. Introduction
Thermoelectric (TE) power generation devices, which directly convert heat into electricity, have long been regarded as the promising next energy revolution since the discovery of the TE effect.1 High-performance TE power generation devices require TE materials with high TE conversion efficiency, which is proportional to the TE figure of merit ZT: ZT = S2σT/κ, where S is the Seebeck coefficient, σ the electrical conductivity, T the absolute temperature, and κ the thermal conductivity including the contributions from electrons (κe) and lattice (κl).2 These concepts indicate that increasing ZT requires both a large power factor PF (PF = S2σ) and a low κ. However, since the Bi2Te3 family was discovered as promising TE materials (ZT ∼ 1),3,4 very few bulk materials have been found with ZT greater than 1, because the phonon mean free path cannot become shorter than the interatomic distance in bulk materials, which results in there being a lower limit to the lattice thermal conductivity.5 Thus, the past advances in ZT are based on nanostructures by reducing the thermal conductivity, improving the ZT larger than 2.1,6–8 Unfortunately, ZT is also nearing a lower limit because the thermal conductivity can not be reduced below the amorphous limit.5 Recently, maximizing the PF has been regarded as another effective approach to realize a new breakthrough in ZT.9For more than decades, improving ZT by maximizing the PF was believed to be a challenge due to the paradox between density-of-states effective mass (m*d) and carrier mobility (μ), which induces a compromise between S and σ.6,10 For a degenerate semiconductor, S is given by:5
 | (1) |
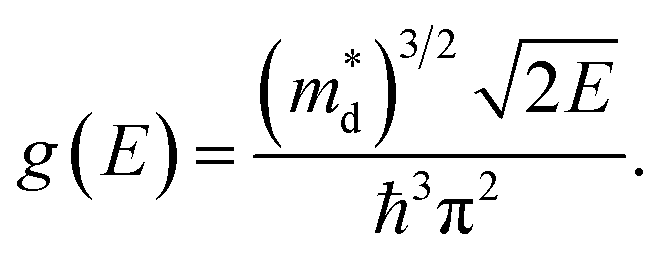 | (2) |
For a given Fermi energy (carrier concentration n), a high overall DOS means a high effective mass (m*d), leading to a high S. Here, the high total DOS can come from either a large number of conducting bands (Nv) or a high band effective mass (m*b) relating to flat bands, which can be related by m*d = Nv2/3m*b.10,11 The number of conducting bands and the flatness of bands (high m*b) usually have different effect on carrier mobility μ. In most cases, the number of conducting bands have little influence on μ unless there is a significant inter-valley scattering. However, μ is low for bands with heavy mass m*b, thus leading to the low electrical conductivity (σ = neμ).10,11 Therefore, increasing band mass is not beneficial to optimize the PF. Band engineering, which can produce a large m*d without reducing μ by increasing the factor Nv, has been used to realize excellent TE performance by maximizing the PF.5,11–13 A doubling of ZT in PbTe above 1.5 was obtained by Heremans et al. in 2008 through distortion of electronic DOS.5 In 2011, Pei et al. realized a high ZT of 1.8 in doped PbTe1−xSex by means of multiple valley degeneracy.11 These breakthroughs indicate that it is feasible to optimize the TE performance by band engineering without using complex nanostructures.
Electronic resonance states and band degeneracy are the two kinds of widely used approaches of band engineering.1,10 Resonant states may occur when dopants have energy levels in band of host materials, which can distort the DOS and increase the effective mass, leading to an enhanced PF.1 Band degeneracy includes orbital degeneracy (the band extrema of multiple bands have their energy within a few kBT of each other) and valley degeneracy (separate pockets of Fermi surface with the same or similar energy).10 A highly degenerate bands lead to an effective increase in Nv, producing an enhanced m*d, and therefore the PF. However, previous improvements are mainly realized in low-dimensional materials. Extending this concept to bulk materials where already exist highly band degenerate would be most useful for rapid integration with applications and has realistic significance. What's more, theoretical prediction and explanation are still lacking. In this work, we improve the room temperature PF of classical bulk thermoelectrics 3 to 8 times by engineering the conduction bands near the Fermi level (multiple bands degeneracy). This is achieved in IV–VI classical bulk semiconductors (PbTe family) without using nanostructures or inducing resonant states by doping. Our results provide an example of searching for optimal PF in classical bulk materials by the characteristic of band degeneracy, which will attract more research interest in optimizing the PF by similar approaches.
2. Computational details
We firstly study the electronic structure of IV–VI semiconductors (PbTe family) by using the full-potential linearized augmented plane-wave method with generalized gradient approximation (GGA) for the exchange–correlation potential, as implemented in the WIEN2K code.14 This method has been successfully used to deal with the electronic structure of various materials.15,16 The convergence results are determined by the cut-off KmaxRmt = 9 for the plane-wave and 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 k points in the first Brillouin zone. The precision of the convergence is set as 10−5 Ry per formula unit. Spin–orbit coupling is included. Then, the electronic transport calculations are done by using the Boltzmann transport theory and relaxation time approximation implemented in the BoltzTraP code.18 Within this approximation, S is independent of relaxation time τ, whereas σ and PF depend linearly on τ. Calculating τ in bulk materials is trivial, which is treated as a constant in the present work. This method has been used for a long time and shows reliable results for TE properties of semiconductors.16,17,19 The classical IV–VI semiconductors belong to the fcc crystal system (rocksalt structure) with the space group Fm
000 k points in the first Brillouin zone. The precision of the convergence is set as 10−5 Ry per formula unit. Spin–orbit coupling is included. Then, the electronic transport calculations are done by using the Boltzmann transport theory and relaxation time approximation implemented in the BoltzTraP code.18 Within this approximation, S is independent of relaxation time τ, whereas σ and PF depend linearly on τ. Calculating τ in bulk materials is trivial, which is treated as a constant in the present work. This method has been used for a long time and shows reliable results for TE properties of semiconductors.16,17,19 The classical IV–VI semiconductors belong to the fcc crystal system (rocksalt structure) with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, and two atoms in the primitive cell individually locate in 4a (Pb) and 4b (Te) sites.20,21
m, and two atoms in the primitive cell individually locate in 4a (Pb) and 4b (Te) sites.20,21
3. Results and discussion
We firstly consider our research for traditional PbTe as shown in Fig. 1(a) and 2(a). Bulk PbTe is known as a traditional p-type TE material.22 Our calculated direct band gap at L point is 0.1 eV, which is smaller than the experimental value of 0.19 eV (ref. 23) due to the gap underestimation in GGA. The conduction bands near the Fermi level are dominated by Pb p states, while valence bands are the Te p states. These states are highly hybridized, and the minimum direct gap appears at L point where the Pb p states shift down due to hybridization with Te d states while the Te p states shift up due to interaction with Pb s states.20 Here, we name the conduction and valence bands near the Fermi level as Pb p-like and Te p-like bands, respectively. The calculated room temperature PFs are ∼1.6 × 1011 W mK−2 s−1 for n-type PbTe and ∼7 × 1011W mK−2 s−1 for p-type PbTe, respectively (the Fermi level shifts up for n-type, while down for p-type). It is thus demonstrated that PbTe is an intrinsic p-type TE material and the p-type PF ∼7 × 1011 W mK−2 s−1 is comparable to ∼6 × 1011 W mK−2 s−1 for p-type Bi2Te3.19 Interestingly, PbTe intrinsically exhibits a trend of multiple bands degeneracy at L point due to the orbits hybridization. However, the degeneracy level is not strong. For example, the energy difference between the band extrema of the first and the second conduction bands near the Fermi level at L point is 1.2 eV, which is far beyond the degeneracy condition (little or even no significant energy difference between the band extrema10). If one can increase the degenerate level of these Pb p-like bands or Te p-like bands, enhanced DOS near the Fermi level will be obtained, and therefore the PF. | ||
| Fig. 1 Calculated band structures and power factors for (a) MTe (M = Pb, Sn, Ge), and (b) MSe (M = Pb, Sn, Ge). Red lines for M = Pb, blue for M = Sn, and magenta for M = Ge. The Fermi level is set at 0 eV. Calculated power factors with n-type above the Fermi-level while p-type under the Fermi-level. Note here that n-type power factors are mainly concerned as the conduction bands are engineered in the substitution. | ||
 | ||
| Fig. 2 Calculated total density of states (DOS) and partial DOS for (a) MTe (M = Pb, Sn, Ge), and (b) MSe (M = Pb, Sn, Ge). Only contributions from s and p states for different atoms are given. Black lines denotes the total DOS, and the partial DOS are represented by the red lines for s states of M, blue lines for p states of M, dark cyan lines for s states of Te and Se, and magenta lines for p states of Te and Se. | ||
Instead of using complex low-dimensions approach, we try atomic substitutions on PbTe in order to check whether an enhanced band degeneracy at L point would be achieved. As shown in Fig. 1(a), we consider atomic substitution at 4a(Pb) site, replacing atom Pb by the atoms in the same group. We represent this first set of materials as MTe (M = Pb, Sn, Ge). As M site going from heavy-mass element to light one, the lattice parameters decrease (6.464 Å for PbTe, 6.327 Å for SnTe and 6.012 for GeTe),24–26 resulting in an increase of interaction between atomic layers and the hybridization of atomic orbits. Consequently, although the Te p-like bands has little changes for we haven't substitute the Te site, we surprisingly find the degeneracy of the M p-like bands at L point is greatly enhanced as going to light-mass element at M site. This increases the room temperature n-type PF from ∼1.6 × 1011 W mK−2 s−1 for PbTe to ∼7.7 × 1011 W mK−2 s−1 for SnTe and ∼12 × 1011 W mK−2 s−1 for GeTe, which enhance about 5 to 8 times. DOS analysis for these compounds indicates that the enhanced PF mostly comes from the increased M p states. We consider in Fig. 2(a) the total and partial DOS of MTe (M = Pb, Sn, Ge), and only s and p states are listed because other states have little contribution to the DOS around the Fermi level. The first conduction band with energy between 0 eV and 1 eV mostly consists of M p states. Although there is an increase of Te p states in the second and third bands, the M p states also make the main contributions (1–3 eV). Thus, the M p states are enhanced as the M p-like bands tend to degenerate at L point, and therefore the DOS and PF. This is achieved without changing the shape of the band structure (band effective mass).
We also consider the second set of materials MSe (M = Pb, Sn, Ge)24,27,28 as shown in Fig. 1(b) and 2(b). Similar enhanced band degeneracy for M p-like bands at L point are found. The room temperature n-type PFs also increase from ∼1.2 × 1011 W mK−2 s−1 for PbSe to ∼4 × 1011 W mK−2 s−1 for SnSe and ∼8 × 1011 W mK−2 s−1 for GeSe, and the M p states also contribute to most of the PF. The PF is increased about 3 to 7 times. However, in comparison with the first set, we find atomic substitution on 4b(Te) site shows opposite effect. Firstly, the energy difference between the band extrema of the first and second Se p like bands at L point is larger than those of Te. And also, the degenerate level of M p like bands is weak this time, leading to a depressed M p states near the Fermi level, and therefore the PF. This is caused by the subdued hybridization of atomic orbits. For example, we have made clear before that the valence Te p states or Se p states shift up at L point due to hybridization with M s states.20 However, as seen in Fig. 2, the energy difference between these two states is expanded as going from Te to Se at 4b site, which results in subdued hybridization and therefore the decreased degenerate level. In both of the two sets, except a sharp DOS for conduction band at around 1 eV, there are also other peaks at higher energies. However, we are willing to realize a low doping level with a carrier concentration about ∼1019 cm−3 in order to give consideration to S and σ.6 In this respect, the paradox between S and σ would usually be represented as carrier concentration.
The two sets of materials we researched both exhibit an enhanced n-type PF by engineering the conduction bands at L point. These bands like small pockets centered at L and mainly associated with M p states, containing electrons for TE transport. The increased degeneracy of M p-like bands leads to an enhanced DOS, and thereby the PF. This is an effective demonstration of the concept of optimizing the PF by band engineering, where the compromise between S and σ is solved, realizing large PF without depressing the carrier concentration and mobility.
We further plot in Fig. 3 the room temperature S, σ, and PFs for the first set of materials MTe (M = Pb, Sn, Ge) as a function of chemical potential, in which we can clearly see how these coefficients vary with different M members. For p-type, these coefficients show little changes for different compounds because of the little change in valence bands dominated by Te p states. The n-type PFs are gradually enhanced as M going from Pb to Ge due to the increased degenerate level in M p-like bands. The enhanced region probably locates in 0.25–0.75 eV, in which the S and σ are simultaneously increased instead of compromising with each other. It is demonstrated that the converged multiple M p-like bands increase the S without overshadowing its σ, and thus realizing in large PFs. Also, as the n-type PFs enhanced, the properties of these thermoelectric materials are changed from p-type to n-type.
 | ||
| Fig. 3 Calculated room temperature Seebeck coefficients, electrical conductivities, and power factors as a function of chemical potential for the first set of materials MTe (M = Pb, Sn, Ge). For n-type, it is found that the Seebeck coefficient and electrical conductivity simultaneously increase instead of compromising with each other, which leads to the enhanced power factors. | ||
4. Conclusions
Motivated by the recent studies that band engineering can solve the paradox between density of states effective mass and carrier mobility, resulting in the increase of the Seebeck coefficient without overshadowing the electrical conductivity, we use the first-principles combined with the Boltzmann transport theory to investigate the electronic structure, Seebeck coefficients S, and power factors PF for traditional semiconductors MTe and MSe. It is found that the room temperature n-type PF of classical thermoelectrics can be increased about 3 to 8 times by engineering the band degeneracy through atom substitution instead of complex low-dimensions approach. This is explained in detail by the calculated band structure and DOS. The present work will inspire further interest in optimizing the TE performance by band engineering.Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant No. 11474113 and 21101123, by the Natural Science Foundation of Hubei Province under Grant No. 2015CFB419, and by the Fundamental Research Funds for the Central Universities under Grant No. HUST: 2015TS019.References
- J. Q. He, M. G. Kanatzidis and V. P. Dravid, Mater. Today, 2013, 16, 166 CrossRef CAS PubMed.
- H. J. Goldsmid, Electronic Refrigeration, Pion, London, 1986, vol. 76 Search PubMed.
- H. J. Goldsmid and R. W. Douglas, J. Appl. Phys., 1954, 5, 386 Search PubMed.
- T. J. Scheidemantel, C. Ambrosch-Draxl, T. Thonhauser, J. V. Badding and J. O. Sofo, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 125210 CrossRef.
- J. P. Heremans, V. Jovovic, E. S. Toberer, A. Saramat, K. Kurosaki, A. Charoenphakdee, S. Yamanaka and G. J. Snyder, Science, 2008, 321, 554 CrossRef CAS PubMed.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105 CrossRef CAS PubMed.
- Y. Gelbstein, J. Davidow, S. Giraid, D. Y. Chung and M. Kanatzidis, Adv. Energy Mater., 2013, 3, 815 CrossRef CAS PubMed.
- D. Wu, et al., J. Am. Chem. Soc., 2014, 136, 11412 CrossRef CAS PubMed.
- D. I. Bilc, G. Hautier, D. Waroquiers, G. M. Rignanese and P. Ghosez, Phys. Rev. Lett., 2015, 114, 136601 CrossRef.
- Y. Pei, H. Wang and G. J. Snyder, Adv. Mater., 2012, 24, 6125 CrossRef CAS PubMed.
- Y. Pei, X. Shi, A. LaLonde, H. Wang, L. Chen and G. J. Snyder, Nature, 2011, 473, 66 CrossRef CAS PubMed.
- J. P. Heremans, B. Wiendlocha and A. M. Chamoire, Energy Environ. Sci., 2012, 5, 5510 CAS.
- C. M. Jaworski, B. Wiendlocha, V. Jovovic and J. P. Heremans, Energy Environ. Sci., 2011, 4, 4155 CAS.
- P. Blaha, K. Schwarz, G. Madsen, D. Kvaniscka, and J. Luitz, Wien2k, An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties, Vienna University of Technology, Vienna, Austria, 2001 Search PubMed.
- G. Y. Gao and K.-L. Yao, Appl. Phys. Lett., 2013, 103, 232409 CrossRef PubMed.
- G. Ding, G. Gao and K. Yao, Sci. Rep., 2015, 5, 9567 CrossRef CAS PubMed.
- L. F. Wan and S. P. Beckman, Phys. Chem. Chem. Phys., 2014, 16, 25337 RSC.
- G. K. H. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67 CrossRef CAS PubMed.
- Q. Shi, Z. Feng, Y. Yan and Y. X. Wang, RSC Adv., 2015, 5, 65133 RSC.
- A. Svane, N. E. Christensen, M. Cardona, A. N. Chantis, M. van Schilfgaarde and T. Kotani, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 245120 CrossRef.
- C. M. I. Okoye, J. Phys.: Condens. Matter, 2002, 14, 8625 CrossRef CAS.
- L. Xu, H. Q. Wang and J. C. Zheng, J. Electron. Mater., 2011, 40, 641 CrossRef CAS.
- Materials - The Landolt-Bornstein Database, ed. O. Madelung, U. Rössler and M. Schulz, Springer, http://www.springermaterials.com.
- P. M. Nikolic, Br. J. Appl. Phys., 1965, 16, 1075 CrossRef CAS.
- R. F. Bis and J. R. Dixon, J. Appl. Phys., 1969, 40, 1918 CrossRef CAS PubMed.
- T. B. Zhukova and A. I. Zaslavskii, Study of the phase transformation, Soviet Physics, Crystallography (=Kristallografiya), 1967, vol. 12, p. 28 Search PubMed.
- A. N. Mariano and K. L. Chopra, Appl. Phys. Lett., 1967, 10, 282 CrossRef CAS PubMed.
- H. Wiedemeier and P. A. Siemers, Z. Anorg. Allg. Chem., 1975, 411, 90 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |