
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsocyanide substitution reactions at the trans labile sites of an iron(II) N-heterocyclic carbene complex†
S.
Haslinger
a,
A. C.
Lindhorst
a,
J. W.
Kück
a,
M.
Cokoja
b,
A.
Pöthig
c and
F. E.
Kühn
*a
aChair of Inorganic Chemistry/Molecular Catalysis, Technische Universität München (TUM), Department of Chemistry/Catalysis Research Center, Lichtenbergstr. 4, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13096
bChair of Inorganic and Organometallic Chemistry, Technische Universität München (TUM), Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany
cCatalysis Research Center, Technische Universität München (TUM), Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany
First published on 30th September 2015
Abstract
A variety of isocyanide-substituted Fe(II) N-heterocyclic carbene (NHC) complexes has been synthesized, starting from an Fe(II) NHC complex with an equatorial, tetradentate bis(pyridyl-NHC) ligand (NCCN). Depending on the relative amount of isocyanide used for the reaction either mono(isocyanide)-substituted or tri(isocyanide)-substituted Fe(II) complexes are obtained. In the case of the tri-substituted complexes single crystal X-ray diffraction reveals the dissociation of one of the pyridyl moieties of the tetradentate NCCN ligand, inducing a meridional, tridentate NCC coordination. As an intermediate for the formation of the tri-substituted complexes a cis di(CNtBu)-substituted Fe(II) complex was identified by NMR spectroscopy. The impact of the isocyanide ligands on the electronic structure of the iron complexes was investigated by cyclic voltammetry, showing an increase in the required potential for the oxidation of Fe(II) to Fe(III) from 423 mV to up to 1092 mV.
Introduction
Iron, the most abundant transition metal, has generated a broad range of chemical applications.1–7 The urge to develop environmentally friendly, cheap, and toxicologically unproblematic catalysts further increased the significance of iron coordination compounds in recent years.8–10 This development in chemical research was mainly driven by studies on biological and bioinspired systems.11–13 With iron active sites playing a crucial role in many biological processes, researchers intended to adapt nature's principles to artificial systems.14 Especially in the field of iron-based, bioinspired oxidation catalysis remarkable advances were made in the last decade, leading to a better understanding of reaction mechanisms and to improved catalyst performance.13,15–17 A common structural motif of these compounds is the use of polydentate ligands with N donor atoms as supporting ligands for Fe(II) and Fe(III) complexes.13,18 Furthermore, the successful implementation of N-heterocyclic carbenes (NHCs) as ligands to a range of transition metal-catalyzed reactions also led to a constantly increasing number of reported iron NHC complexes with catalytic activity.8,19–22 Electronic fine-tuning for an improved catalytic performance can be achieved by either variation of the polydentate ligand or by introduction of ligands with specific donor or acceptor properties into accessible coordination sites.23 For instance, the impact of axial ligands with diverse electronic properties on the oxygenation reactivity in apical positions has been investigated in some detail.24,25Our group, for example, developed an Fe(II) oxidation catalyst bearing a tetradentate, equatorial bis(pyridyl-NHC) ligand (complex 1, Fig. 1).26 Complex 1 catalyzes the oxidation of unreactive alkanes, aromatic hydroxylation, and olefin epoxidation.27–29 Beside modification of the equatorial tetradentate ligand it was demonstrated that the electronic structure of 1 is significantly influenced by axial ligand exchange.30–32 Phosphine- and pyridine-based ligands were used in particular to probe the impact on the redox behavior.30
 | ||
| Fig. 1 Fe(II) complexes 1 and 2a with a tetradentate, equatorial bis(pyridyl-NHC) ligand. 1 has been applied as a catalyst for the oxidation of unreactive alkanes, aromatic hydroxylation, and olefin epoxidation.27–292a as catalyst for the oxidation of unreactive alkanes showed an improved performance compared to 1.29 | ||
In order to broaden the scope, tert-butyl isocyanide as easy-to-handle ligand with both pronounced π-acceptor and σ-donor abilities was successfully used to create an Fe(II) catalyst (2a) with only one accessible coordination site.29 As 2a shows an improved performance as catalyst in the oxidation of unreactive alkanes, it was intended to expand the number of mono(isocyanide)-substituted Fe(II) complexes derived from 1. The use of isocyanides as ligands, being isolobal to carbon monoxide (CO), is well established in coordination chemistry.33–37 Modification of the substitution pattern at the isocyanide functionality allows modulation of electronic and steric properties, rendering it a useful alternative to CO.33,38,39
In this article the syntheses, structural, and electronic characterization of Fe(II) complexes with four isocyanide ligands bearing different substituents are described. The range of mono(isocyanide)-substituted derivatives of 1 is extended and also the behavior of 1 towards an excess of the respective isocyanide is investigated. Reaction monitoring by NMR spectroscopy is used to identify possible intermediates during the course of reaction. Finally, the impact of isocyanide ligands of the electronic structure of the obtained Fe(II) complexes is studied by cyclic voltammetry and the results are discussed in the context of previously reported findings on the electronic tuning of Fe(II) complexes.30
Results and discussion
Syntheses of mono(isocyanide)-substituted complexes
For substitution experiments with different isocyanides the starting complex 1 was dissolved in acetonitrile. Addition of isocyanide at room temperature resulted in a fast change of color from the characteristic orange of 1 to bright yellow. Four different isocyanides were used in this study: tert-butyl isocyanide (CNtBu), cyclohexyl isocyanide (CNCy), benzyl isocyanide (CNBn), and 4-methoxyphenyl isocyanide (CN-p-PhOMe). Previously, we reported the reaction of 1 with a slight excess (1.0 to 1.5 equiv.) of CNtBu,29 leading to the formation of mono(CNtBu)-substituted complex 2a which was isolated after 30 min reaction time at room temperature (Scheme 1). | ||
| Scheme 1 Syntheses of mono(isocyanide)-substituted complexes 2a–5a and tri(isocyanide)-substituted complexes 2b–5b starting from 1. Complex 2a had already been reported before by our group.29tBu = tert-butyl, Cy = cyclohexyl, Bn = benzyl, p-PhOMe = 4-methoxyphenyl. | ||
The same synthetic procedure was now applied to the reactions of 1 with CNCy, CNBn, and CN-p-PhOMe. In all three cases, the respective mono(isocycanide)-substituted complexes 3a, 4a, and 5a were isolated in yields between 59% and 94%. In the 1H NMR spectra of Fe(II) complexes bearing an equatorially coordinating tetradentate NCCN ligand, the chemical shift of the ortho proton signals of the pyridyl moieties are highly sensitive to changes of the electronic structure of the complex, e.g., caused by axial ligand exchange.26,30 For the starting material 1 the signal is observed at 9.57 ppm, while it is found at 9.26 ppm for 2a. 1H NMR spectroscopy of 3a, 4a, and 5a reveal the signals of the ortho protons at 9.26 ppm, 9.28 ppm, and 9.23 ppm, respectively. These shifts are almost identical for all four mono(isocyanide)-substituted Fe(II) complexes, indicating a negligible influence of the substituents of the isocyanide functional group on the electronic structure of the respective complexes. Only minor changes in the chemical shifts of all other signals of the NCCN ligand are found in the 1H NMR spectra. Characteristic for the monosubstitution pattern, the signal of the CH2 bridge of the NCCN ligand changes from a singlet in case of 1 to a doublet of doublets for 2a–5a. This is a consequence of broken symmetry in the equatorial plane, as the trans-positioned axial ligands are no longer identical. Additionally, ESI-MS data confirmed the monosubstitution in solution (m/z 611.47 (3a), 660.41 (4a), 676.41 (5a)).
Single crystals suitable for X-ray diffraction were obtained for 3a and 4a by slow diffusion of diethyl ether into a saturated acetonitrile solution of the respective complexes. The molecular structures of the cationic fragments are shown in Fig. 2. The molecular structures of 3a and 4a are very similar with the tetradentate NCCN ligand being arranged equatorially.
 | ||
| Fig. 2 ORTEP style representation of the cationic fragments of 3a and 4a with ellipsoids shown at a 50% probability level. Hydrogen atoms and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: (3a) Fe1–C1 1.831(4), Fe1–C10 1.836(4), Fe1–N3 2.087(3), Fe1–N6 2.088(3), Fe1–C18 1.847(4), Fe1–N8 1.959(3), C18–N7 1.153(4), N3–Fe1–N6 115.03(11), C18–Fe1–N8 172.82(14); (4a) Fe1–C1 1.839(3), Fe1–C10 1.834(3), Fe1–N3 2.079(2), Fe1–N6 2.086(2), Fe1–C18 1.852(3), Fe1–N8 1.953(2), C18–N7 1.152(3), N3–Fe1–N6 115.11(8), C18–Fe1–N8 170.56(10). | ||
Compared to both starting material 1 and mono(CNtBu)-substituted complex 2a the distances between the Fe atom and the coordinating atoms in the equatorial plane remain almost unchanged for 3a and 4a. Relevant structural parameters for the coordinated isocyanides such as the Fe–C distances (Fe1–C18: 1.847(4) Å for 3a and 1.852(3) Å for 4a) and the C–N bond lengths (C18–N7: 1.153(4) Å for 3a and 1.152(3) Å for 4a) are identical within the error margin for both mono(isocyanide)-substituted complexes. The distance of the acetonitrile ligand trans to the isocyanide is slightly larger for 2a–4a (Fe1–N8: 1.963(4) Å for 2a,29 1.959(3) Å for 3a, and 1.953(2) Å for 4a) compared to 1 (1.9151(1) Å).
Syntheses of tri(isocyanide)-substituted complexes
Based on the successful isolation of the monosubstituted Fe(II) complexes the synthesis of disubstituted derivatives with both axial acetonitrile ligands being replaced by isocyanides was targeted. Thus, the amount of isocyanide for the reactions with 1 was increased to 2 equiv. on NMR scale, starting with CNtBu. Unexpectedly, 1H NMR spectroscopy revealed a mixture of the monosubstituted complex 2a and a second compound. Based on the signal pattern this compound could not be described as the expected disubstituted Fe(II) complex. In order to identify the second species, an excess of CNtBu (5 equiv.) was used for the reaction on an NMR scale. The resulting 1H NMR spectrum revealed complete conversion of 1 to the second, unidentified species and the presence of free CNtBu (Fig. 3). An additional increase of the amount of CNtBu to 10 equiv. did not result in any further change of the 1H NMR spectrum. | ||
| Fig. 3 Selected range (5.5 ppm to 10 ppm) of 1H NMR spectra of the reaction of 1 with 2 equiv. CNtBu and 5 equiv. CNtBu after 18 hours reaction time. For comparison, 1H NMR spectra of isolated 1, 2a, and 2b are shown. | ||
Based on the splitting pattern of the signals and the integral ratios the novel compound was identified as the tri(CNtBu)-substituted Fe(II) complex 2b (see Scheme 1). One of the pyridyl moieties of the former tetradentate NCCN ligand decoordinates the iron atom, and the additionally accessible coordination site is occupied by CNtBu trans to an NHC moiety. Also, both axial acetonitrile ligands have been replaced by CNtBu. As a consequence of the broken symmetry a full set of signals, with no symmetry equivalent protons, is observed for the NCCN ligand in the 1H NMR spectrum of 2b. This is quite different from the situation observed for 1 and 2a–5a. With chemical shifts of 8.75 ppm and 8.33 ppm both signals of the ortho protons of the pyridyl moieties are shifted upfield compared to 9.26 ppm for 2a and 9.57 ppm for 1.26,30 An additional confirmation of the trisubstitution pattern is given by ESI-MS data, showing a distinct signal at m/z 751.82.
The tri(isocyanide) substitution was successfully extended to the isocyanides CNCy, CNBn, and CN-p-PhOMe to yield complexes 3b, 4b, and 5b, respectively. In all cases, an excess of the respective isocyanide in the presence of 1 leads to the formation of the tri(isocyanide)-substituted products. The course of the reaction is very similar to the formation of 2b from 1, with an excess of isocyanide being required for selective formation of 2b–5b. Addition of only 2 equiv. isocyanide yields a mixture of the respective mono- and tri-substituted complexes. The presence of three isocyanides coordinating to Fe is confirmed by ESI-MS in solution (m/z 829.94 (3b), 354.37 (4b, dicationic fragment z = 2), 901.65 (5b)). 1H NMR spectroscopy reveals signals in a very similar pattern to 2b for all three complexes with the signal of the ortho protons of the pyridyl moieties showing chemical shifts of 8.77 ppm and 8.32 ppm for 3b, 8.56 ppm and 8.20 ppm for 4b, and 8.53 ppm and 8.44 ppm for 5b (compare 2b: 8.75 ppm and 8.33 ppm). In addition to characterization in solution, single crystals suitable for X-ray diffraction were obtained for 2b, 3b, and 5b (Fig. 4).
 | ||
| Fig. 4 ORTEP style representation of the cationic fragments of 2b, 3b, and 5b with ellipsoids shown at a 50% probability level. Hydrogen atoms and PF6− counter ions are omitted for clarity. Selected bond lengths [Å] and angles [°]: (2b) Fe1–C1 1.885(4), Fe1–C10 1.952(4), Fe1–N3 2.069(3), Fe1–C18 1.904(4), Fe1–C23 1.881(4), Fe1–C28 1.892(4), C18–N7 1.149(5), C23–N8 1.152(5), C28–N9 1.152(5), N3–Fe1–C18 92.69(14), C10–Fe1–C18 101.41(16), C23–Fe1–C28 175.25(16); (3b) Fe1–C1 1.869(4), Fe1–C10 1.951(4), Fe1–N3 2.060(3), Fe1–C18 1.890(5), Fe1–C25 1.892(4), Fe1–C32 1.892(5), C18–N7 1.159(5), C25–N8 1.151(5), C32–N9 1.144(5), N3–Fe1–C18 94.60(15), C10–Fe1–C18 100.30(17), C25–Fe1–C32 176.06(17); (5b) Fe1–C1 1.8825(19), Fe1–C10 1.9460(19), Fe1–N3 2.0594(16), Fe1–C18 1.881(2), Fe1–C26 1.893(2), Fe1–C34 1.876(2), C18–N7 1.158(3), C26–N8 1.154(3), C34–N9 1.161(3), N3–Fe1–C18 93.87(7), C10–Fe1–C18 100.23(8), C26–Fe1–C34 176.89(8). | ||
From the molecular structures shown in Fig. 4 it is evident that indeed one of the pyridyl moieties is no longer coordinating to the Fe atom as the 1H NMR data suggest. The former tetradentate NCCN ligands coordinate meridional in a tridentate fashion and three isocyanides occupy the remaining coordination sites, completing the octahedral geometry of the Fe(II) complexes. Due to the induced asymmetry, the Fe–CNHC bond lengths differ between the two NHC units. Within the meridional tridentate NCC ligands, the central NHC units show bond lengths of Fe1–C1 1.885(4) Å, 1.869(4) Å, and 1.8825(19) Å for 2b, 3b, and 5b, respectively. Compared to that, the Fe1–C10 bonds of the other NHC units are slightly longer with values of 1.952(4) Å, 1.951(4) Å, and 1.9460(19) Å. The distances between iron and the isocyanide ligands trans to the NHC moiety are almost identical for 2b, 3b, and 5b with 1.904(4) Å, 1.890(5) Å, and 1.881(2) Å, respectively, and with a range of 1.876(2) Å to 1.892(5) Å for the two isocyanides trans to each other. In all cases the isocyanide C–N bond lengths are in the typical range of isocyanide-coordinated Fe–NHC complexes with values in the range of 1.144(5) Å to 1.161(3) Å.40 Interestingly, no well-defined difference is observed between the isocyanide trans to the NHC unit and the isocyanides that are positioned trans to each other.
Time-dependent analysis of ligand exchange reaction
During the formation of the tri(isocyanide)-substituted Fe(II) complexes it is likely that an intermediate species bearing two isocyanide ligands is formed. As characteristic signals of 2a and 2b in 1H NMR do not overlap (see Fig. 3), the reaction of 1 with 5 equiv. CNtBu was monitored over time by 1H NMR spectroscopy. With the first data point being collected after 3 minutes, the initial reaction step (formation of 2a from 1) proceeded too fast to be monitored at room temperature. Thus, the spectra were recorded at 10 °C. Based on the 1H NMR spectra, four different species were observed during the reaction (Fig. 5).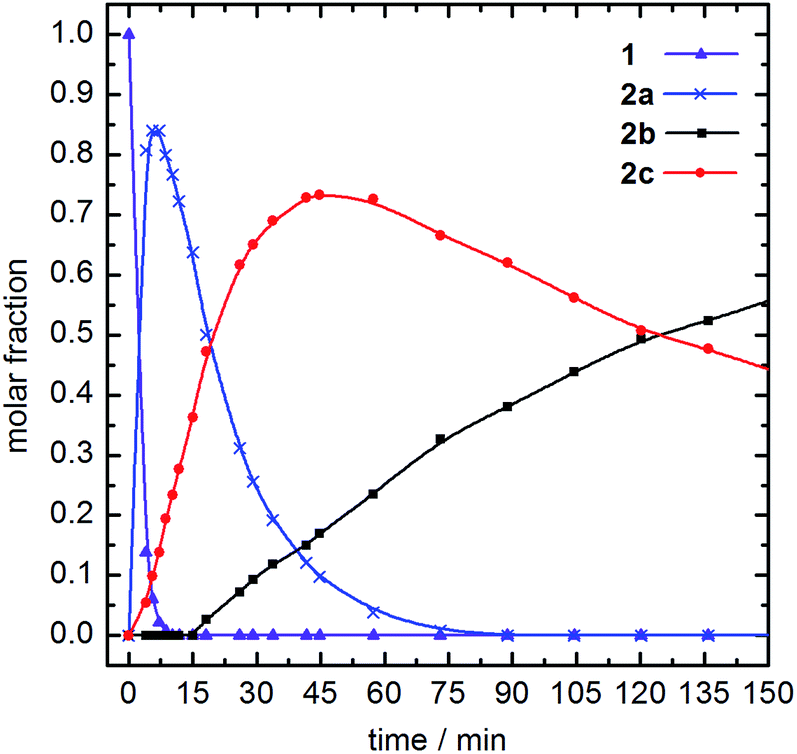 | ||
| Fig. 5 Time-dependent analysis of the reaction of 1 (purple triangles) with 5 equiv. CNtBu at 10 °C, monitored by 1H NMR spectroscopy. Initial formation of 2a (blue cross) is observed, followed by the formation of an additional, intermediate compound (red circles) and 2b (black squares). | ||
The amount of starting material 1 (purple triangles) quickly decays after addition of CNtBu. At the first data point after 3 minutes only 14% of 1 are left in the reaction mixture, and after 9 min complex 1 has been converted completely to its isocyanide-substituted derivatives. As initial intermediate mono(CNtBu)-substituted complex 2a is formed (blue ×) with a peak accumulation of 84% after 7 min. Subsequently, 2a further reacts to form its higher-substituted derivatives and after 80 min 2a is no longer detected in the reaction mixture. Most interestingly, at the first data point after 3 min a third species beside 1 and 2a is identified (red circles) which accumulates to 73% after 40 min before its amount starts to decrease again. Based on the signal pattern in 1H NMR, this intermediate is not identical to 1, 2a, or the tri(CNtBu)-substituted 2b, which starts to form after 15 min (black squares). The integral ratio and the splitting pattern of the intermediate species suggest a di(CNtBu)-substituted complex 2c. Two singlet signals at 1.09 ppm and 1.42 ppm with each having an integral of 9 are assigned to the two tert-butyl groups of the isocyanide ligands while the signal of the CH2 bridge is split up to a doublet of doublets at 6.49 ppm with an overall integral of 2. As a full set of signals is observed for the remaining protons of the tetradentate ligand, a highly asymmetric structure for 2c is likely (Scheme 2). The observed asymmetry can only be achieved by a cis arrangement of the CNtBu ligands, as a simple exchange of the trans labile acetonitrile of 2a would result in a highly symmetric structure with an 1H NMR signal pattern similar to 1.30 Hence, the meridional coordination pattern shown in Scheme 2 is indicated by the spectroscopic data for 2c as well. Based on time-dependent ratios of the Fe(II) complexes as shown in Fig. 5 each subsequent substitution by one additional CNtBu starting from 1 proceeds slower. In the presence of an excess of isocyanide complex 1 is bound to form 2b, which is the thermodynamically most favored compound in the substitution equilibrium. The thermodynamic stability of 2b and all other tri-substituted complexes 3b–5b is also evident from 1H NMR data in acetonitrile, as the respective spectra do not change after storing the samples for several days under air. No exchange of the coordinating isocyanides by acetonitrile is observed, which stands in contrast to the observations reported for phosphine-based ligands.30
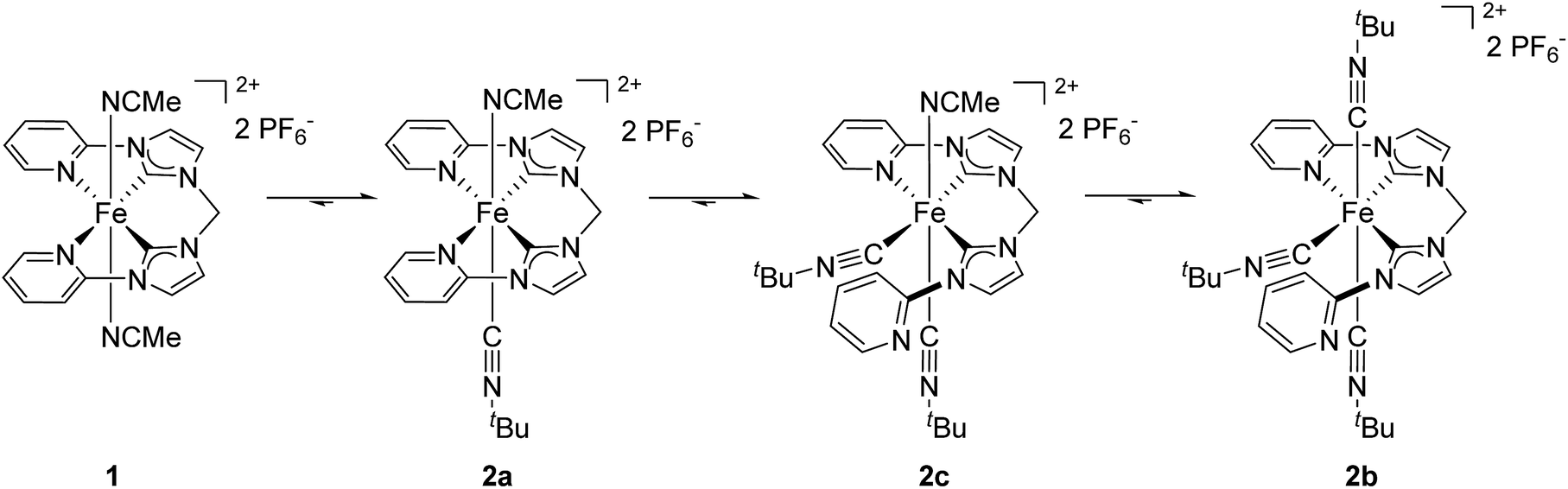 | ||
| Scheme 2 Suggested pathway for the reaction of 1 with excess of CNtBu to form tri(CNtBu)-substituted complex 2b. | ||
Electrochemical investigations
Previous studies have revealed a significant influence of the substitution of the labile acetonitrile ligands of 1 on the electronic structure of Fe(II) complexes.30 This was investigated both theoretically by DFT calculations and experimentally by determination of the half-cell potentials with cyclic voltammetry. While the half-cell potential of 1 was determined to be 423 mV (versus Fc/Fc+), an exchange of only one acetonitrile ligand with PMe3 reduced the half-cell potential to 325 mV. For the mono(isocyanide)-substituted complexes 2a–5a an increase in half-cell potential is expected compared to 1, as isocyanides are π-acceptor ligands in addition to their σ-donor properties. Coordination of a π-acceptor ligand should result in a decreased electron density at the iron atom, resulting in a higher potential required for the oxidation of Fe(II) to Fe(III). This should also hold true for the tri(isocyanide)-substituted complexes 2b–5b. In order to investigate the impact of the isocyanide ligands on the redox behavior of the respective complexes, all compounds 2–5 were subjected to cyclic voltammetry experiments and the respective data is shown in Fig. 6. | ||
| Fig. 6 Cyclic voltammograms of mono(isocyanide)-(2a–5a, left) and tri(isocyanide)-substituted Fe(II) complexes (2b–5b, right). | ||
For all mono(isocyanide)-substituted complexes 2a–5a a fully reversible oxidation, assigned to the Fe(II)/Fe(III) redox couple, is observed. The half-cell potentials versus Fc/Fc+ are 539 mV for 2a, 540 mV for 3a, 557 mV for 4a, and 573 mV for 5a. As expected, in all cases the required potential for the oxidation of Fe(II) is significantly higher (116 mV to 150 mV) than observed for 1 and also for its mono(PMe3)-substituted derivative reported earlier. Within the series of 2a–5a, the variation of the substituents on the isocyanide functionalities has a small influence on the half-cell potentials. Bearing alkyl substituents, complexes 2a (tBu) and 3a (Cy) show identical half-cell potentials, while the benzyl substituent in case of 4a leads to a slightly increased potential. For 5a this effect is more pronounced as the aryl substituent (p-PhOMe) influences the isocyanide functionality to a greater extent due to π-conjugation. Cyclic voltammetry experiments of tri(isocyanide)-substituted complexes 2b–5b, however, lead to different results. In all cases, the oxidation is no longer reversible and requires a significantly higher potential than 2a–5a. The peak current for the oxidation from Fe(II) to Fe(III) was found at 994 mV for 2b, 1091 mV for 3b, 1076 mV for 4b, and 1092 mV for 5b (all versus Fc/Fc+). As 2b–5b are bearing three isocyanide ligands, the higher oxidation potentials can be explained by the accumulated electron-withdrawing ability of the π-acceptor ligands, leading to a significantly reduced electron density at the iron atoms. A possible explanation for the irreversibility is the instability of the oxidized Fe complexes in the oxidation state +III, induced by the presence of three π-acceptor ligands.
It is likely that after oxidation to Fe(III), the remaining electron density is no longer sufficient to maintain the given coordination environment and therefore one of the isocyanide ligands is removed from the coordination sphere. Consequently, the loss of one π-acceptor ligand most likely changes the respective reduction potential and the overall redox process is no longer reversible. Indeed, an independent, irreversible reduction wave was observed for 2b (592 mV), 3b (487 mV), and 4b (562 mV). In case of 5b this reduction wave is indicated only vaguely at 448 mV. Based on the NMR data for the reaction of 1 with excess CNtBu to form 2b (see above) and the irreversibility of the electrochemical oxidation of 2b–5b from Fe(II) to Fe(III), these reduction waves are understood to have their origin in the respective di(isocyanide)-substituted intermediates.
Experimental
General remarks
All chemicals were purchased from commercial suppliers and used without further purification. Complexes 1 and 2a were synthesized according to the literature.26,29,41 Liquid NMR spectra were recorded on a Bruker Avance DPX 400 and a Bruker Ultrashield 500 Plus with cryo unit. Chemical shifts are given in parts per million (ppm) and the spectra were referenced by using the residual solvent shifts as internal standards (MeCN-d3, 1H NMR δ 1.94, 13C NMR δ 1.32). A Thermo Scientific LCQ/Fleet spectrometer by Thermo Fisher Scientific was used to collect MS-ESI data and elemental analyses were obtained from the microanalytical laboratory of TUM.Single crystal X-ray diffraction
Single crystals of 3a, 4a, 2b, and 3b suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into an acetonitrile solution of respective compounds. In case of 5b slow diffusion of diethyl ether into an acetone solution of 5b yielded single crystals. 3a: yellow, monoclinic crystal system, space group P21/c (no. 14), a = 19.1865(9) Å, b = 10.7359(4) Å, c = 15.8045(7) Å, β = 40.406(2)°, V = 3211.7(2) Å3. 4a: yellow, triclinic crystal system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 8.0698(5) Å, b = 10.1378(7) Å, c = 19.5360(13) Å, α = 98.549(3)°, β = 101.122(3)°, γ = 100.397(3)°, V = 1514.35(17) Å3. 2b: yellow, triclinic crystal system, space group P (no. 2), a = 12.0501(4) Å, b = 17.2210(5) Å, c = 20.7696(6) Å, α = 69.987(2)°, β = 86.127(2)°, γ = 85.440(2)°, V = 4033.2(2) Å3. 3b: yellow, monoclinic crystal system, space group P21/c (no. 14), a = 19.9697(7) Å, b = 11.5746(3) Å, c = 18.9375(7) Å, β = 100.129(2)°, V = 4309.0(2) Å3. 5b: yellow, monoclinic crystal system, space group P21/c (no. 14), a = 8.08910(10) Å, b = 21.8620(3) Å, c = 24.9410(3) Å, β = 96.7210(10)°, V = 4380.35(10) Å3. Crystallographic data for structures 3a, 4a, 2b, 3b, and 5b have been deposited with the Cambridge Crystallographic Data Centre (CCDC 1422720–1422724†).
(no. 2), a = 8.0698(5) Å, b = 10.1378(7) Å, c = 19.5360(13) Å, α = 98.549(3)°, β = 101.122(3)°, γ = 100.397(3)°, V = 1514.35(17) Å3. 2b: yellow, triclinic crystal system, space group P (no. 2), a = 12.0501(4) Å, b = 17.2210(5) Å, c = 20.7696(6) Å, α = 69.987(2)°, β = 86.127(2)°, γ = 85.440(2)°, V = 4033.2(2) Å3. 3b: yellow, monoclinic crystal system, space group P21/c (no. 14), a = 19.9697(7) Å, b = 11.5746(3) Å, c = 18.9375(7) Å, β = 100.129(2)°, V = 4309.0(2) Å3. 5b: yellow, monoclinic crystal system, space group P21/c (no. 14), a = 8.08910(10) Å, b = 21.8620(3) Å, c = 24.9410(3) Å, β = 96.7210(10)°, V = 4380.35(10) Å3. Crystallographic data for structures 3a, 4a, 2b, 3b, and 5b have been deposited with the Cambridge Crystallographic Data Centre (CCDC 1422720–1422724†).
Cyclic voltammetry
A PalmSens EmStat3+ potentiostat was used together with the PSTrace4 software for cyclic voltammetry measurements. A platinum wire was chosen as counter electrode, together with glassy carbon as working electrode and Ag/AgNO3 (0.01 M in acetonitrile with 0.1 M tetrabutylammonium hexafluoro-phosphate) as reference electrode. For all measurements, 3.0 mg of the respective compound was dissolved in 1.0 mL of acetonitrile under inert conditions, containing 0.1 mmol tetrabutylammonium hexafluorophosphate. The potential was scanned with 100 mV s−1versus Ag/AgNO3 and the obtained values were referenced versus the Fc/Fc+ redox couple as internal standard (0.48 V versus SCE).42Syntheses of complexes
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) 2157. MS-ESI (m/z): [3a–2PF6]2+ calcd, 233.58; found, 233.44. Anal. calcd for C26H28F12FeN8P2: C, 39.12; H, 3.54; N, 14.04. Found: C, 39.01; H, 3.34; N, 13.69.
N) 2173. MS-ESI (m/z): [4a–PF6]+ calcd, 661.11; found, 660.41; [4a–2PF6]+ calcd, 258.08; found, 257.92. Anal. calcd for C27H24F12FeN8P2: C, 40.22; H, 3.00; N, 13.90. Found: C, 39.80; H, 3.30; N, 13.81.
N) 2122. MS-ESI (m/z): [5a–PF6]+ calcd, 677.11; found, 676.41; [5a–MeCN–PF6]+ calcd, 636.08; found, 635.43.
N) 2150. MS-ESI (m/z): [2b–PF6]+ calcd, 752.25; found, 751.82.
N) 2168. MS-ESI (m/z): [3b–PF6]+ calcd, 830.29; found, 829.94. Anal. calcd for C38H47F12FeN9P2: C, 46.78; H, 4.86; N, 12.92. Found: C, 47.16; H, 4.71; N, 12.76.
N) 2177. MS-ESI (m/z): [4b–2PF6]2+ calcd, 354.62; found, 354.37. Anal. calcd for C41H35F12FeN9P2: C, 49.27; H, 3.53; N, 12.61. Found: C, 48.98; H, 3.56; N, 12.47.
N) 2135. MS-ESI (m/z): [5b–PF6]+ calcd, 902.19; found, 901.95.
N) 2157. MS-ESI (m/z): [3a–2PF6]2+ calcd, 233.58; found, 233.44. Anal. calcd for C26H28F12FeN8P2: C, 39.12; H, 3.54; N, 14.04. Found: C, 39.01; H, 3.34; N, 13.69.
N) 2173. MS-ESI (m/z): [4a–PF6]+ calcd, 661.11; found, 660.41; [4a–2PF6]+ calcd, 258.08; found, 257.92. Anal. calcd for C27H24F12FeN8P2: C, 40.22; H, 3.00; N, 13.90. Found: C, 39.80; H, 3.30; N, 13.81.
N) 2122. MS-ESI (m/z): [5a–PF6]+ calcd, 677.11; found, 676.41; [5a–MeCN–PF6]+ calcd, 636.08; found, 635.43.
N) 2150. MS-ESI (m/z): [2b–PF6]+ calcd, 752.25; found, 751.82.
N) 2168. MS-ESI (m/z): [3b–PF6]+ calcd, 830.29; found, 829.94. Anal. calcd for C38H47F12FeN9P2: C, 46.78; H, 4.86; N, 12.92. Found: C, 47.16; H, 4.71; N, 12.76.
N) 2177. MS-ESI (m/z): [4b–2PF6]2+ calcd, 354.62; found, 354.37. Anal. calcd for C41H35F12FeN9P2: C, 49.27; H, 3.53; N, 12.61. Found: C, 48.98; H, 3.56; N, 12.47.
N) 2135. MS-ESI (m/z): [5b–PF6]+ calcd, 902.19; found, 901.95.
Conclusions
Addition of substituted isocyanides to a solution of the Fe(II) NHC complex 1 with trans labile sites leads to the syntheses of isocyanide-substituted Fe(II) complexes 3a–5a and 2b–5b. tBu, Cy, Bn, and p-PhOMe were used as substituents on the isocyanide moiety. Characterization of the products by NMR spectroscopy and single crystal X-ray diffraction (SC-XRD; for 3a, 4a, 2b, 3b, and 5b) reveals mono(isocyanide) substitution or tri(isocyanide) substitution depending on the relative amount of isocyanide used for the syntheses. Isolation of di(isocyanide)-substituted products is not possible, as the addition of 2 equiv. isocyanide to 1 results in a mixture of mono- and tri-substituted complexes. The molecular structures of the tri-substituted complexes obtained by SC-XRD show meridional coordination of the three isocyanides and a change from an equatorial, fourfold coordination to a meridional, threefold coordination of the polydentate NCCN ligand. 1H NMR spectroscopy was applied to monitor the reaction of 1 with an excess of CNtBu (5 equiv.). Within 40 min an intermediate species is amounting to 73% before reacting to the tri-substituted complex 2b. Based on the NMR data the intermediate can be identified as di(isocyanide)-substituted derivative 2c of complex 1. In 2c one pyridyl moiety of the NCCN ligand has been removed from the coordination sphere and a cis coordination of the two isocyanide ligands is observed. Cyclic voltammetry experiments on all compounds reveal a significantly increased half-cell potential for the fully reversible redox step Fe(II)/Fe(III) of mono-substituted complexes 2a–5a (up to 573 mV versus Fc/Fc+) compared to starting complex 1 (423 mV). The tri-substituted complexes 2b–5b required a further increase in potential up to 1092 mV for the oxidation to Fe(III), albeit this redox step is found to be irreversible. The presented structural and analytical data can be used as a starting point for further modification of the Fe(II) complexes by adjusting the isocyanide substituents in terms of steric demand or σ-donor/π-acceptor ratio.Acknowledgements
The authors gratefully acknowledge support by the TUM graduate school. A. C. L. further appreciates the financial support by the Fonds der Chemischen Industrie (FCI). S. H. thanks Tassilo M. F. Restle for his valuable contributions.References
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS PubMed.
- S. Blanchard, E. Derat, M. Desage-El Murr, L. Fensterbank, M. Malacria and V. Mouriès-Mansuy, Eur. J. Inorg. Chem., 2012, 376–389 CrossRef CAS.
- K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC.
- A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 1364–1367 CrossRef PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed.
- K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2364 CrossRef CAS PubMed.
- E. C. Theil and D. J. Goss, Chem. Rev., 2009, 109, 4568–4579 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- A. B. McQuarters, M. W. Wolf, A. P. Hunt and N. Lehnert, Angew. Chem., Int. Ed., 2014, 53, 4750–4752 CrossRef CAS PubMed.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- W. Nam, Y. Ryu and W. Song, JBIC, J. Biol. Inorg. Chem., 2004, 9, 654–660 CrossRef CAS PubMed.
- K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73–96 CrossRef CAS.
- S. Bang, S. Park, Y.-M. Lee, S. Hong, K.-B. Cho and W. Nam, Angew. Chem., Int. Ed., 2014, 53, 7843–7847 CrossRef CAS PubMed.
- B. J. Wallar and J. D. Lipscomb, Chem. Rev., 1996, 96, 2625–2658 CrossRef CAS PubMed.
- E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS.
- D. J. Nelson, Eur. J. Inorg. Chem., 2015, 2012–2027 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- Y. Hitomi, K. Arakawa and M. Kodera, Chem.–Eur. J., 2013, 19, 14697–14701 CrossRef PubMed.
- T. Chantarojsiri, Y. Sun, J. R. Long and C. J. Chang, Inorg. Chem., 2015, 54, 5879–5887 CrossRef CAS PubMed.
- S. Bandyopadhyay, A. Rana, K. Mittra, S. Samanta, K. Sengupta and A. Dey, Inorg. Chem., 2014, 53, 10150–10158 CrossRef CAS PubMed.
- A. Raba, M. Cokoja, S. Ewald, K. Riener, E. Herdtweck, A. Pöthig, W. A. Herrmann and F. E. Kühn, Organometallics, 2012, 31, 2793–2800 CrossRef CAS.
- A. Raba, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Commun., 2014, 50, 11454–11457 RSC.
- J. W. Kück, A. Raba, I. I. E. Markovits, M. Cokoja and F. E. Kühn, ChemCatChem, 2014, 6, 1882–1886 CrossRef.
- S. Haslinger, A. Raba, M. Cokoja, A. Pöthig and F. E. Kühn, J. Catal., 2015, 331, 147–153 CrossRef CAS.
- S. Haslinger, J. W. Kück, E. M. Hahn, M. Cokoja, A. Pöthig, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2014, 53, 11573–11583 CrossRef CAS PubMed.
- J. Rieb, A. Raba, S. Haslinger, M. Kaspar, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2014, 53, 9598–9606 CrossRef CAS PubMed.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed.
- A. E. Carpenter, C. C. Mokhtarzadeh, D. S. Ripatti, I. Havrylyuk, R. Kamezawa, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2015, 54, 2936–2944 CrossRef CAS PubMed.
- H.-K. Kim, J.-H. Lee, Y.-J. Kim, Z. Nu Zheng and S. W. Lee, Eur. J. Inorg. Chem., 2013, 4958–4969 CAS.
- L. Weber, Angew. Chem., Int. Ed., 1998, 37, 1515–1517 CrossRef CAS.
- Y. Yamamoto, Coord. Chem. Rev., 1980, 32, 193–233 CrossRef CAS.
- F. A. Cotton and F. Zingales, J. Am. Chem. Soc., 1961, 83, 351–355 CrossRef CAS.
- M. V. Barybin, Coord. Chem. Rev., 2010, 254, 1240–1252 CrossRef CAS.
- L. A. Labios, M. D. Millard, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2009, 131, 11318–11319 CrossRef CAS PubMed.
- J. J. Scepaniak, R. P. Bontchev, D. L. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2011, 50, 6630–6633 CrossRef CAS PubMed.
- A. Raba, M. R. Anneser, D. Jantke, M. Cokoja, W. A. Herrmann and F. E. Kühn, Tetrahedron Lett., 2013, 54, 3384–3387 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Listings of NMR data. CCDC 1422720–1422724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra18270k |
| This journal is © The Royal Society of Chemistry 2015 |