
Overview of the effect of monomers and green solvents on thermoresponsive copolymers: phase transition temperature and surface properties†
P. Madhusudhana Reddy‡
a,
Chi-Jung Chang*a,
Shih-Rong Hsieh‡b,
Hsin-Chun Huanga and
Ming-Ching Leeb
aDepartment of Chemical Engineering, Feng Chia University, 100, Wenhwa Road, Seatwen, Taichung 40724, Taiwan, Republic of China. E-mail: changcj@fcu.edu.tw; Tel: +886-4-24517250 ext. 3678
bDepartment of Surgery, Taichung Veterans General Hospital, 1650 Taiwan Boulevard Sect. 4, Taichung 40705, Taiwan, Republic of China
First published on 8th October 2015
Abstract
The present study explores the effect of the monomer substitution pattern of different copolymers on their volume phase transition temperature (VPTT) and surface wetting properties (SWPs) with the aid of differential scanning calorimetry (DSC), contact angle (CA), scanning electron microscopy (SEM) and Fourier transform-infrared spectroscopy (FT-IR) measurements. The present experimental results unveil that the VPTT and SWPs were greatly governed by the ability of the neighboring side chains to form intramolecular hydrogen bonds and the hydrophobic–hydrophilic balance of the polymer. The substitution of some of the acrylic acid (AA) monomers in the copolymer hydrogel P(NIPAM-co-AA) by the polyethyleneglycolmethacrylate (PEGMA) monomer resulted in the copolymer hydrogel, P(NIPAM-co-PEGMA-co-AA). The PEGMA in P(NIPAM-co-PEGMA-co-AA) can promote the intermolecular hydrogen bonds of the copolymer with the water molecules and thus hinders the formation of the intramolecular hydrogen bonds. Consequently, the VPTT of the P(NIPAM-co-PEGMA-co-AA) occurs at higher temperatures when compared to the P(NIPAM-co-AA) and P(NIPAM-co-PEGMA) systems. Further, in view of the potential applications of ionic liquids (IL) in polymer science, the effect of IL on the VPTT and SWP of the copolymers was investigated. The knowledge from this study can pave the way to engineer stimuli responsive polymers for a wide range of applications in the modern era.
1. Introduction
Hydrogels (HGs) have been intensively investigated owing to their appealing physicochemical properties which are similar to those of living tissues, for instance, high water content, controllable porosity, mechanical properties and composition.1 Further, the plethora of applications of these hydrogels is diverse as they can serve as controlled drug delivery vehicles for therapeutics and as scaffolds for promoting tissue regeneration.2–4 Thermosensitive poly(N-isopropylacrylamide) (PNIPAM) based materials have attracted considerable attention among researchers in the scientific world.5–7 PNIPAM based hydrogel in water is highly swollen at low temperature, but undergoes a sharp phase transition at the volume phase transition temperature (VPTT) and is thus shrunken.8 Further heating of the hydrogel in water may lead to the formation of aggregates after the VPTT.9 Developing strategies for controlling the VPTT and polymeric assembly is a significant research topic for diverse fields of science. This research holds potentials in the areas of drug delivery, biological sensing and imaging, tissue engineering and removal of dye from water.9–12 In this regard, besides the common approach of introducing either hydrophilic or hydrophobic block into parent chain,13 there are other potential methods in use to tune the VPTT of hydrogels by changing the quality of solvent medium through external stimuli such as pH, ionic strength, light, and electric field.14,15Nowadays, there is a growing interest in the development of ionic liquids (ILs) among the researchers across the scientific world because they are not any longer just esoteric compounds, but are proved to be the valuable and useful candidates in a lot of different applications.16–21 Recently, ILs have been used in polymer related research.22–26 For instance, ILs can be used as a stabilizing agents to control the average size and surface area of polymer beads due to their profound surface-active properties.23 Besides, these ILs can also be used as a regulators to control the phase transitions of thermoresponsive polymers.24–26
Thermoresponsiveness of polymers indeed dependent on the nature of the monomers or blocks involved in the structure of the polymers.27 Polymer with more hydrophilic monomers shows higher phase transition temperature, while polymer with more hydrophobic monomers exhibits phase transition at relatively lower temperature.28 As mentioned in earlier discussion, the phase transition temperature can also be varied by the addition of different kinds of additives including ionic solid, surfactant, urea, and sugar as external stimuli into the polymer in water.29–33 However, unlike these traditional additives, ILs have some unique properties, such as negligible vapor pressure, tunable viscosity, strong polarity, high ionic conductivity properties, chemical stability, miscibility with water and organic solvents, and good thermal stability (some ILs are exempted from high thermal stability).34,35 Furthermore, ILs offer a great flexibility in their properties since the possible combinations of cations and anions are quite high. Therefore, it is more convenient and desirable to use these ILs to tune the phase transition temperature of polymers.
In spite of tremendous applications of ILs in polymer science,22–26 limited research has been devoted to tune the phase transition temperature of thermoresponsive polymers in water by the addition of ILs.24,27,36,37 However, these few studies focusing on a single polymer with a transition temperature close to body temperature. To the best of our knowledge, no single study has been carried out to explore the effects of ILs on VPTT of polymeric HG with various degree of hydrophilicity. Hence, it is of our prime interest to answer the most exciting open question that how the VPTTs and surface wetting properties of synthetic polymeric HGs is influenced with changing hydrophilicity/hydrophobicity. Further, the changes in these properties were also studied in response to the type of aqueous IL.
In the present work, the VPTTs and surface wetting properties (SWPs) of poly(N-isopropylacrylamide-co-polyethyleneglycolmethacrylate) P(NIPAM-co-PEGMA), poly(N-isopropylacrylamide-co-acrylic acid) P(NIPAM-co-AA), poly(N-isopropylacrylamide-co-polyethyleneglycolmethacrylate-co-acrylic acid) P(NIPAM-co-PEGMA-AA) were studied. The VPTT of P(NIPAM-co-PEGMA-AA) copolymer was observed at higher temperature when compared to that of P(NIPAM-co-PEGMA) and P(NIPAM-co-AA). The higher VPTT of P(NIPAM-co-PEGMA-AA) was attributed to the intermolecular hydrogen bonds with surrounded water molecules and thus led to the formation of hydration layer around the polymer backbone. Therefore, this hydration layer can increase the solubility of the polymer and, thus consequently P(NIPAM-co-PEGMA-AA) exhibits higher VPTT. In contrast to this, the close spatial arrangement of hydrogen bond accepting group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and hydrogen bond donating group (–NH–) in P(NIPAM-co-PEGMA) and P(NIPAM-co-AA) induces intramolecular hydrogen bonding, and therefore these polymers exhibited phase transition at lower temperatures. Further, the effect of ILs on the VPTTs and SWPs of polymers was found to be dependent on the ability of the polymers to form the hydrogen bonds.
O) and hydrogen bond donating group (–NH–) in P(NIPAM-co-PEGMA) and P(NIPAM-co-AA) induces intramolecular hydrogen bonding, and therefore these polymers exhibited phase transition at lower temperatures. Further, the effect of ILs on the VPTTs and SWPs of polymers was found to be dependent on the ability of the polymers to form the hydrogen bonds.
2. Experimental
2.1 Materials
The monomers N-isopropylacrylamide (NIPAM), and acrylic acid (AA) were provided by Acros. N,N,N′,N′-Tetramethylethylenediamine (TEMED), ammonium persulfate (APS), N,N′-methylenebisacrylamide (MBAA) (cross-linker), and polyethyleneglycolmethacrylate (PEGMA) were received from the Aldrich chemical co. The ILs, [Bmim][Cl], [Bmim][BF4] and [Bmim][HSO4] were obtained from the Uniregion Bio-Tech Corp. Deionized (DI) water with a resistivity of 18.2 MΩ cm was obtained from Roda purification system (Te Chen Co. Ltd.) and used for all the samples preparation. All the chemicals were used as received. All the prepared hydrogel samples were lyophilized using a freeze-dryer before the measurements.2.2 Synthesis of P(NIPAM-co-AA) and P(NIPAM-co-PEGMA) hydrogels
P(NIPAM-co-AA) hydrogel was prepared by using the procedure reported in the previous reports.25,38 Briefly, NIPAM, AA and MBAA dissolved in phosphate buffered saline. The reaction mixture was stirred for 30 min in a three-necked round-bottom flask equipped with a condenser under a N2 purge. Subsequently, to this solution, TEMED and APS were added and the resultant solution was allowed to stir for 19 h at room temperature, under a N2 atmosphere. The prepared hydrogel was cleaned with DI water to remove unreacted monomer and cross-linker. The synthesized hydrogel was characterized by the FT-IR spectroscopy. The absence of carbon–carbon double bond has confirmed the complete polymerization of monomers, NIPAM and AAc. The same procedure has been used for the preparation of P(NIPAM-co-PEGMA) hydrogel with the replacement of AA by PEGMA. The feed ratio of [NIPAM]/[PEGMA]/[APS]/[MBAA] in PG is 9.54 × 10−5/3.75 × 10−5/3.94 × 10−7/2.36 × 10−7 mol L−1, respectively and the feed ratio of [NIPAM]/[AA]/[APS]/[MBAA] in P(NIPAM-co-AA) is 9.54 × 10−5/3.75 × 10−5/3.94 × 10−7/2.36 × 10−7 mol L−1, respectively.2.3 Synthesis of P(NIPAM-co-PEGMA-co-AA) hydrogels
The synthesis of P(NIPAM-co-PEGMA-co-AA) hydrogels was as follows. Initially, PEGMA was mixed with AA thoroughly. Subsequently, the resultant solution was transferred into a three-necked round-bottom flask containing NIPAM and MBAA in phosphate buffered saline. The reaction mixture was lasted for 30 min with stirring and continuous N2 gas purging. Finally, to this solution TEMED and APS were added and reacted for 19 h at room temperature under nitrogen atmosphere. The feed ratio of [NIPAM]/[PEGMA]/[AA]/[APS]/[MBAA] in P(NIPAM-co-PEGMA-co-AA)-4 is 9.54 × 10−5/1.87 × 10−5/1.87 × 10−5/3.94 × 10−7/2.36 × 10−7 mol L−1, respectively and the feed ratio of [NIPAM]/[PEGMA]/[AA]/[APS]/[MBAA] in P(NIPAM-co-PEGMA-co-AA)-7 is 9.54 × 10−5/2.25 × 10−5/1.5 × 10−5/3.94 × 10−7/2.36 × 10−7 mol L−1, respectively. FT-IR spectroscopy was employed for characterization of the prepared hydrogel. All the hydrogels were freeze-dried before the use. The chemical structures of P(NIPAM-co-AA), P(NIPAM-co-PEGMA) and P(NIPAM-co-PEGMA-co-AA) hydrogels were shown in Fig. 1. The chemical compositions of all the HGs and their corresponding VPTTs (°C), CA (degree) and porosity were shown in Table 1.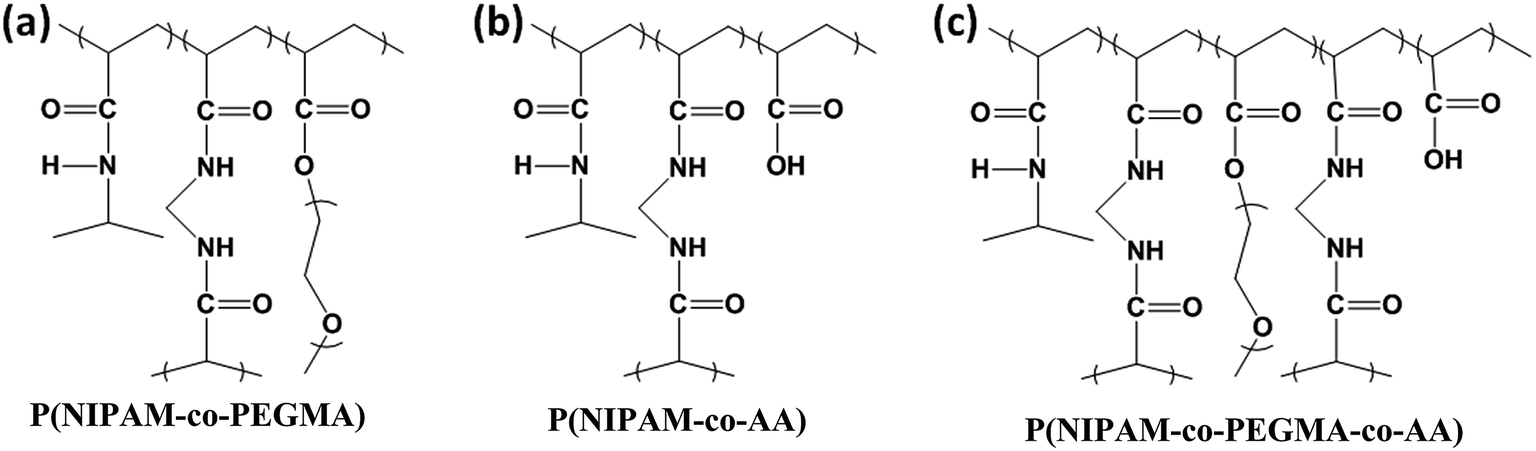 | ||
| Fig. 1 Chemical structures of (a) P(NIPAM-co-PEGMA), (b) P(NIPAM-co-AA) and (c) P(NIPAM-co-PEGMA-co-AA) hydrogels. | ||
| HG | Without IL | [Bmim][BF4] | [Bmim][Cl] | [Bmim][HSO4] |
|---|---|---|---|---|
| a PL = large pores; PS = small pores; PN = no pores. | ||||
| P(NIPAM-co-PEGMA | 40.8/103/PL | 47.7/83/PL | 43.5/72/PS | —/110/PN |
| P(NIPAM-co-AA) | 42.3/69/PL | 49.9/42/PL | 46.5/45/PL | 30/76/PN |
| P(NIPAM-co-PEGMA-co-AA)-4 | 62.6/48/PL | 60/72/PN | 55/77/PS | 38.6/95/PN |
| P(NIPAM-co-PEGMA-co-AA)-7 | 59.7/55/PL | 54.3/60/PN | 48.3/61/PN | 40.6/102/PN |
2.4 Sample preparation
Aqueous solutions of polymer hydrogels (0.5% w/v) were prepared for DSC measurements by following the procedure, with minimal modifications, reported in the previous literature.39 Appropriate amount of freeze-dried hydrogel was mechanically ground. This mechanically ground HG was dissolved in sufficient amount of DI water and stirred for four days to get the required polymer concentration (0.5% w/v). After stirring, the samples were additionally sonicated for 30 min. Then, certain amount of IL was added to the above-mentioned solution.2.5 Differential scanning calorimetry (DSC) measurements
TA-MDSC-2920 (TA Instruments, New Castle, DE, USA) instrument was used for the differential scanning calorimetry measurements sample solutions. All the experiments were performed in a nitrogen atmosphere. Aluminum pans were used for calorimetry measurements. DSC scans were recorded at a scan rate of 5 °C min−1 with a pre-scan thermostat of 5–10 minutes to ensure thermal equilibrium. The uncertainties in temperature and heat flow readings are ±0.02 °C and ±0.1%, respectively. The transition temperature was defined as the initial break point of DSC thermogram.2.6 Contact angle (CA) measurements
Contact angles of aqueous solution of IL on the surfaces of dry polymer hydrogels were determined using a contact angle meter (CAM-100, Creating-Nanotech Co.). Dry polymer hydrogel films were prepared by freeze drying method. The average CA was obtained using the software Euresys (Easy Grab Multicam for Picolo Version 3.8.3). In a typical measurement, a fixed volume of sample droplet (water having IL) was placed on the flat surface of dry hydrogel and started obtaining CA after allowing drop to held in place for ca. 5 s. The CA measurements are made on both the left and right sides of the droplet, and the mean CA is reported. The images of the solution droplets were captured by a coupled device camera at room temperature.2.7 Scanning electron microscopy (SEM)
SEM micrographs were obtained on the field emission scanning electron microscope (FESEM) by an energy dispersive X-ray (EDX) with a HITACH S-4800 FESEM. The specimens for SEM micrographs were prepared from dry polymer hydrogel films with and without aqueous IL treatment. Dry hydrogel films were immersed in aqueous IL solution for 30 s. Finally, these aqueous IL solution treated sample specimens were subjected to freeze drying and used for SEM images.2.8 Fourier transform infrared spectroscopy (FTIR) measurements
FTIR spectra for all the HGs in the absence and presence of ILs were obtained by using a Nicolet Protégé -460 FTIR spectrophotometer equipped with a variable-temperature sample holder. Each IR spectrum reported here was an average of 32 scans using a spectral resolution of 4 cm−1 for an acceptable signal-to-noise ratio.3. Results and discussion
PNIPAM hydrogel undergoes volume phase transition (VPT) at VPTT of ∼32 °C.8 The introduction of hydrophilic monomers can increase the VPTT of PNIPAM based hydrogels, while reverse is true when hydrophobic monomers are used.40 Fig. 2 illustrates the VPTT values, of the hydrogels in water, extracted from the DSC thermograms. At low temperatures all the HGs possess hydration layers. As the temperature increases, the hydration layer will be disturbed through increased hydrophobic interactions and this leads to the phase transition which can be evidenced from the DSC thermograms (Fig. 2). The VPTTs of all HGs were given in Table 1. From Fig. 2, it is clear that the P(NIPAM-co-PEGMA), P(NIPAM-co-AA), P(NIPAM-co-PEGMA-co-AA)-4 and P(NIPAM-co-PEGMA-co-AA)-7 HGs in water have shown the phase transitions at ∼40.8, 42.3, 62.6 and 59.7 °C, respectively. | ||
| Fig. 2 DSC thermograms for the polymeric hydrogels (0.5% w/v) in water; P(NIPAM-co-PEGMA) (a), P(NIPAM-co-AA) (b), P(NIPAM-co-PEGMA-AA)-4 (c) and P(NIPAM-co-PEGMA-AA)-7 (d). | ||
The P(NIPAM-co-PEGMA-co-AA)-4 has shown the maximum VPTT value when compared to that of remaining HGs in water. The elevated VPTT of P(NIPAM-co-PEGMA-co-AA)-4 is attributed to the formation of strong intermolecular hydrogen bonds by the well separated neighborhood of the repeating units of NIPAM, PEGMA and AA of the copolymer system with the surrounded water molecules. These strong intermolecular hydrogen bonds between repeating units of the copolymer and water molecules make the hydration cluster around the polymer chain and thereby, more heat is required to disturb this cluster. It leads to the higher VPTT. The PEGMA in P(NIPAM-co-PEGMA-co-AA)-4 can increase the bulkiness of the polymer chain, and thereby, restricts the flexibility of the entire copolymer system and consequently hinders the formation of the intramolecular hydrogen bonds. In contrast to this, the intra molecular hydrogen bonds were expected for P(NIPAM-co-AA) system as neighborhood of the repeating units of NIPAM and AA arranged closely in the copolymer system and leads to a strong increase of the intramolecular (CO⋯HN) hydrogen bond. The dominated intra molecular hydrogen bonds make the polymer to become insoluble at lower temperature during heating process and thus exhibited lower VPTT. The copolymer hydrogel, P(NIPAM-co-PEGMA) consists of NIPAM and PEGMA as repeating units. NIPAM consists of hydrogen-donor (–NH) and hydrogen-acceptor (CO) functionalities, contrary to PEGMA which provides only hydrogen-acceptor functionalities. When compared AA with PEGMA, PEGMA can offer an additional hydrophobic alkyl chain portion. Because of this additional hydrophobic portion and lack of hydrogen bond donating group in PEGMA, P(NIPAM-co-PEGMA) has exhibited further lower VPTT value when compared to P(NIPAM-co-AA).
Interestingly, with further enhancement of PEGMA content as in P(NIPAM-co-PEGMA-co-AA)-7, the VPTT was observed at lower temperature than that of P(NIPAM-co-PEGMA-co-AA)-4. From these results, one can understand that the copolymer hydrogel can exhibit phase transition at higher temperature, when monomers present in specific ratio. Similar kind of results were observed in the literature.41,42 According to Cai et al.41 the LCST was reduced when the hydrophilic acrylic acid content was increased in the copolymer. The LCST of the poly(N,N-di-ethylacrylamide-co-acrylic acid) poly(DEAAm-AA) was increased with the addition of AA in relatively small amount (7%). However, further enhancement of AA content (>7%) in DEAAm-AA copolymer lead to decrease the LCST as compared to pure PDEAAM. When, acrylic acid content reaches 25%, the polymer became insoluble in water. Further, Richtering et al.42 have observed that an unusual dependence of transition temperature on the composition of the copolymer microgels consisting of N,N-diethylacrylamide-co-N-isopropylacrylamide (PDEAAM-co-PNIPAM). These results indicated that the polymer undergo hydrophobic collapse due to the strong hydrogen bonding at certain composition.
Since ILS can influence the transition temperature of thermoresponsive polymers,24,25,27,36,37 we firstly examined the effect of [Bmim][BF4] and [Bmim][Cl] on the VPTTs of the copolymer HGs.
It is obvious from Fig. S1 (ESI†) that the VPTTs of P(NIPAM-co-PEGMA) and P(NIPAM-co-AA) increased upon the addition of [Bmim][BF4] and [Bmim][Cl]. On the other hand, the VPTTs of the P(NIPAM-co-PEGMA-co-AA) systems decreased with the addition of [Bmim][BF4] and [Bmim][Cl]. This difference in the behavior of ILs towards VPTTs of polymers can be explained tentatively as follows. [Bmim][BF4] can form four hydrogen bonds with water molecules through four hydrogen bonding acceptors, four fluorine atoms.43 It is reasonable to expect that the availability of free water molecules from the bulk water to BF4 ions is sufficiently enough as the solubility of P(NIPAM-co-AA) and P(NIPAM-co-PEGMA) is relatively low when compared to that of P(NIPAM-co-PEGMA-co-AA) systems. As a result, the additional heat is required to break the hydrogen bonds between F and water molecule and thus the P(NIPAM-co-AA) and P(NIPAM-co-PEGMA) systems have shown the elevated VPTTs in the presence of the ILs. The similar argument can be applied to explain the effect of [Bmim][Cl]. Conversely, P(NIPAM-co-PEGMA-co-AA) in H2O solution did not supply enough number of water molecules to the F− and Cl− as large portion of the bulk water was involved in the construction of water cluster around the polymer. As a result, F− and Cl− ions start to make the hydrogen bonds with the water molecules in the hydration layer of P(NIPAM-co-PEGMA-co-AA), which resulted in the breaking of hydrogen bonds between P(NIPAM-co-PEGMA-co-AA) polymer and water molecules due to the strong hydrogen bond accepting nature of F− and Cl−. Consequently, the P(NIPAM-co-PEGMA-co-AA) systems have exhibited phase transition at lower temperatures in the presence of [Bmim][BF4] and [Bmim][Cl]. On the basis of the discussion described above and in order to understanding of the mechanism more intuitively, we present a schematic illustration in Scheme 1.
 | ||
| Scheme 1 Schematic illustration of the behavior of P(NIPAM-co-PEGMA) or P(NIPAM-co-AA) and P(NIPAM-co-PEGMA-co-AA) in water in the presence of [Bmim][BF4]. | ||
Unlike BF4− and Cl−, HSO4− ion has produced the same effect on the all systems. All the HGs have shown the reduced VPTT in the presence of HSO4− (Fig. S1†). This indicates that HSO4− is the most effective ion in destabilizing the hydrated structures of all the HGs. Further, the decrease in VPTTs of all the HGs in the presence of [Bmim][HSO4] was attributed to the increase in intermolecular and intramolecular hydrogen bonds between the IL-water and polymer complex. According to the previous report,44 the unordered zones in bulk water become more ordered with the addition of a highly ordered anion such as HSO4− by making the hydrogen bonds with water molecules. The addition of ordered HSO4− ions to the HGs in water can break the hydrogen bonds between polymer and water and consequently form the new hydrogen bonds between IL and water. This consequent breaking and making of hydrogen bonds lead to the reduced VPTT in the presence of [Bmim][HSO4].
The distinct changes in surface wetting properties of copolymers in response to changes in solute identity can be monitored through the CA measurements. These CA measurements can provide valuable information regarding the behavior of dry polymer surfaces and thereby these materials can be used effectively as smart materials.45,46 Fig. 3 provides the alterations in CA measurements of various aqueous ILs solutions on the surfaces of different dry HGs films. The CA measurements of deionized water on the surfaces of HGs films were also included for the sake of comparison. As shown in Fig. 3, the deionized water has shown maximum and minimum CA values on P(NIPAM-co-PEGMA) (103°) and P(NIPAM-co-PEGMA-co-AA)-4 (48°) hydrogel surfaces respectively, indicating that P(NIPAM-co-PEGMA) and P(NIPAM-co-PEGMA-co-AA)-4 systems were more hydrophobic and hydrophilic, respectively. The reduced hydrophilic nature of P(NIPAM-co-AA) system can be attributed to the dominating intramolecular hydrogen bonds among the AA chains.
 | ||
| Fig. 3 Changes in contact angles of various aqueous solutions of ILs on the dry surfaces of HGs. The concentration of IL was chosen as 1 M. | ||
The solute (ILs) induced wettability changes of HGs can be extracted from the changes in CA as shown in Fig. 3. Being consistent with our DSC results, aqueous [Bmim][BF4] or Bmim][Cl] have shown the lower CA on P(NIPAM-co-PEGMA) (83° for [Bmim][BF4], 52° for [Bmim][Cl]) and P(NIPAM-co-AA) (42° for [Bmim][BF4] and 45° for [Bmim][Cl]) HGs surfaces than that of DI water. These results indicate that the switching of dry P(NIPAM-co-PEGMA) or P(NIPAM-co-AA) hydrogel surfaces from hydrophobic (in the presence of water) to hydrophilic (in the presence of aqueous [Bmim][BF4] or [Bmim][Cl]). In contrast to this, P(NIPAM-co-PEGMA-co-AA) HGs surfaces switch from hydrophilic (in the presence of water) to hydrophobic (in the presence of [Bmim][BF4] or Bmim][Cl]) as CA values increased for aqueous [Bmim][BF4] or Bmim][Cl] solution. The differences in CA of same aqueous ILs solutions ([Bmim][BF4] or Bmim][Cl]) on different HGs are resulted from the variation of chemical compositions of HGs. However, the aqueous solution of [Bmim][HSO4] has shown increased CA values than that of DI water on all the HGs in spite of various chemical composition of HGs. This indicates that the surfaces of all the HGs can behave hydrophobic in response to [Bmim][HSO4] (kosmotrope), as HSO4− is known for the salting out mechanism.47
Our prior CA measurements have uncovered that the type of anion has a great influence in changing the surface behavior of HGs.25 In order to gain deeper knowledge on the influence of varying anion identity in altering the surface morphology of dry hydrogels, further we have collected the SEM images of dry HGs in the absence and presence of ILs. SEM images of all the dry HGs without and with the treatment of 1 M ILs were shown in Fig. 4.
 | ||
| Fig. 4 SEM images of aqueous ILs treated dry hydrogels. The concentration of IL was chosen as 1 M. | ||
All the four HGs have shown the porous structure in the absence of ILs. However, this porous structure was disappeared for the P(NIPAM-co-PEGMA-co-AA) systems when treated with aqueous [Bmim][BF4]. In contrast to this, P(NIPAM-co-PEGMA) and P(NIPAM-co-AA) systems have retained their porous structures even after being treated with aqueous [Bmim][BF4]. These results indicated that the BF4− ions can act as water structure makers for P(NIPAM-co-PEGMA) and P(NIPAM-co-AA) systems, while water structure breakers for P(NIPAM-co-PEGMA-co-AA) systems by collapsing porous structure. In the presence of Cl−, the surface of the dried for P(NIPAM-co-PEGMA) and P(NIPAM-co-AA) systems films remain porous, however, the pores turned smaller. In addition, the surfaces of for P(NIPAM-co-PEGMA-co-AA)-4 and P(NIPAM-PEGMA-AA)-7 became porous with small pores and nonporous, respectively, in response to Cl−. HSO4− has exhibited water structure breaker behavior for all the HGs, as all the HGs have lost their porous structure after being treated with aqueous [Bmim][HSO4].
The PNIPAM based HGs were characterized by the help of FTIR spectroscopy. The FTIR spectra of all the HGs were shown in the Fig. 5. However, the spectrum of P(NIPAM-co-PEGMA-co-AA)-7 was not shown as it has shown almost similar spectrum to P(NIPAM-co-PEGMA-co-AA)-4. As shown in the Fig. 5, the peak at 3200–3400 cm−1 was assigned to the N–H stretching vibrations in NIPAM. The peaks at 1630 and 1540 cm−1 were attributed to the amide I and amide II bands, respectively. The amide I and II regions were mainly attributed from the CO and N–H groups, respectively. D2O was used as a solvent instead H2O for the FTIR measurements. The major advantage of using D2O as a solvent was preventing the overlap of the –OH band of water molecules at about 1630 cm−1 with amide I band of HGs and facilitates the analysis of the hydration state of the amide CO groups of HGs.48,49 To evaluate the influence of ILs on the VPTT of HGs, the changes in amide I were mainly considered as these peaks were very sensitive to hydrogen bonding.48–50
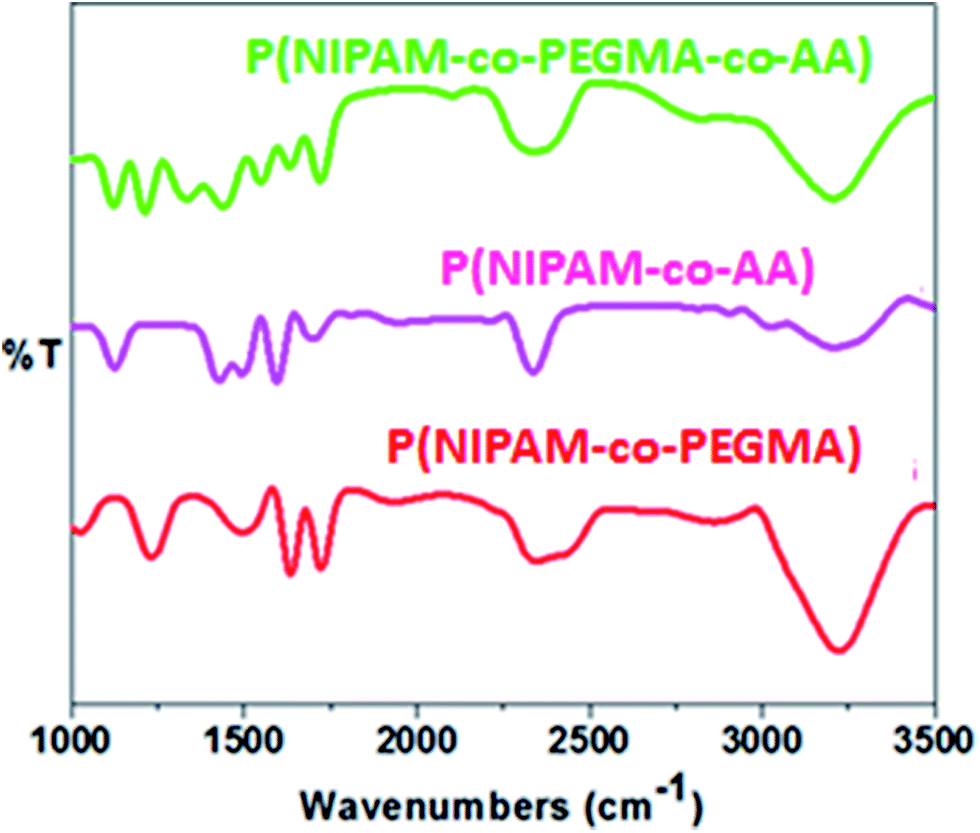 | ||
| Fig. 5 FTIR spectra of P(NIPAM-co-PEGMA), P(NIPAM-co-AA) and P(NIPAM-co-PEGMA-co-AA) HGs in D2O. | ||
FTIR measurements of P(NIPAM-co-PEGMA) and P(NIPAM-co-PEGMA-co-AA)-4 systems were carried out at temperature well below to the VPTT (VPTT-10 °C) in order to evaluate the effect of ILs and presented in Fig. 6. Since, these measurements were performed at temperature well below to the VPTT, the alterations in amide peaks were attributed to the addition of ILs solely. As shown in Fig. 6(a), the position of amide I peak shifted to the lower wavenumber and this can be an evidence for the existence of intermolecular hydrogen bonds (CO⋯D–O–D) between polymer and D2O in the presence of ILs. This indicated that the solubility of HG was not disturbed with the addition of ILs. On the other hand, the amide I peak at ∼1630 cm−1 was significantly shifted to higher wavenumbers with addition of IL as shown in Fig. 6(b). This symbolizes that the addition of IL into the P(NIPAM-co-PEGMA-co-AA)-4 + D2O system can weaken the intermolecular hydrogen bonds between CO⋯D–O–D and consequently strengthen the intramolecular hydrogen bonds (CO⋯H–N) among the polymer chains. The similar kind of shifts in the position of peaks relating to the amide bands were observed due to the dehydration when the polymer solution was heated.48 Thus, the present FT-IR results unveil that the IL act as structure stabilizers for P(NIPAM-co-PEGMA) system, while act as structure destabilizers for P(NIPAM-co-PEGMA-co-AA) system. Further, from these results one can also understand that the behavior of IL depends on the chemical composition of the polymer. Indeed, the peak at ∼1630 cm−1 in P(NIPAM-co-PEGMA) system could shift to higher wavenumbers with the addition of [Bmim][HSO4]. However, we have not measured the FTIR spectra of P(NIPAM-co-PEGMA) system in the presence of [Bmim][HSO4] due to immediate formation of precipitation. For the sake of brevity, we have not given the spectra for P(NIPAM-co-AA) and P(NIPAM-co-PEGMA-co-AA)-7 systems.
 | ||
| Fig. 6 FTIR spectra of (a) P(NIPAM-co-PEGMA) and (b) P(NIPAM-co-PEGMA-co-AA)-4 polymers in the presence of different ILs below the phase transition temperature (VPTT-10 °C). | ||
To understand the temperature effect on the phase transition behavior of HGs, we have measured the FTIR spectra at temperature well above to the VPTT (VPTT + 5 °C). As shown in Fig. 7(a) and (b), the amide I peaks shifted from ∼1630 cm−1 to higher wavenumbers upon heating. This reflects that both the HGs undergo phase transition due to dehydration although they differ in hydrophilicity. This shift in amide I peak towards higher wavenumber was due to the transformation of local environment of the CO groups from hydrophilic to hydrophobic as the hydrated structure of polymer collapsed. In addition, the number and strength of intra molecular hydrogen bonds increased when compare to the intermolecular hydrogen bonds. This behavior leads to a rearrangement of the chains in the cross linked hydrogel during the phase transition.51 At elevated temperatures, i.e. well above to the VPTT, the effect of ILs on swollen/shrinking of HGs became weak and temperature effect dominated. This can be demonstrated from the FTIR spectra of P(NIPAM-co-PEGMA) and P(NIPAM-co-PEGMA-co-AA)-4 in the presence and absence of ILs at elevated temperatures as shown in Fig. 7. As shown in this figure, the amide I peak shifted to higher wavenumbers irrespective of type of IL. Nevertheless, in addition to temperature effect, the phase transition of HGs can be amplified by the addition of IL having water structure breaker ion.
 | ||
| Fig. 7 FTIR spectra of (a) P(NIPAM-co-PEGMA) and (b) P(NIPAM-co-PEGMA-co-AA)-4 polymers in the presence of different ILs above the phase transition temperature (VPTT + 5 °C). | ||
4. Conclusions
In this work DSC, CA, SEM and FTIR measurements were employed to elucidate the role of chemical composition on the VPTT of HGs in water. Further, the influence of ILs on VPTT and surface properties of HGs were studied. P(NIPAM-co-PEGMA-AA)-4 exhibited phase transition at higher temperatures than that of P(NIPAM-co-AA) or P(NIPAM-co-PEGMA). On the basis of our experimental results, we infer that the PEGMA in P(NIPAM-co-PEGMA-co-AA) can promote the intermolecular hydrogen bonds of the copolymer with the water molecules and thus hinders the formation of the intramolecular hydrogen bonds. Consequently, the VPTT of the P(NIPAM-co-PEGMA-co-AA) occurs at higher temperatures as compared to the P(NIPAM-co-AA) and P(NIPAM-co-PEGMA) systems. The P(NIPAM-co-PEGMA-co-AA) systems have exhibited significant shrinking in response to BF4− ion, while the reverse is true for P(NIPAM-co-PEGMA) or P(NIPAM-co-AA) system. However, despite the variations in chemical compositions, all the HGs have shown the shrinking and aggregating properties in response to HSO4− ion due to its strong “water structure breaker” behavior. Further, the effects of ILs on VPTT of HGs were well correlated with the surface wettability and morphology. Thus, the understanding of behavior of VPTT and surface properties of PNIPAM based HGs in the presence of various ILs may be helpful for the control over tuning the VPTT and thereby polymeric assemblies. In the future, the finding about the effect of ILs on volume phase transition temperature (VPTT) and swelling/shirking behavior of HGs in water can contribute to facilitate the application of HGs in drug delivery. ILs induced changes in surface morphology of dried HGs films may help to develop the scaffolds for promoting tissue regeneration.Acknowledgements
This work is supported by Ministry of Science and Technology in Taiwan under contract number NSC102-2632-E-035-001-MY3. The authors also thank the financial support from Feng Chia University and the Taichung Veterans General Hospital under contract number TCVGH-FCU1048201. The authors appreciate the Precision Instrument Support Center of Feng Chia University for providing the measurement facilities.Notes and references
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- T. M. O'Shea, A. A. Aimetti, E. Kim, V. Yesilyurt and R. Lange, Adv. Mater., 2015, 27, 65–72 CrossRef PubMed.
- A. H. van Hove, M. J. G. Beltejar and D. S. W. Benoit, Biomaterials, 2014, 35, 9719–9730 CrossRef CAS PubMed.
- M. K. Nguyen and E. Alsberg, Prog. Polym. Sci., 2014, 39, 1235–1265 CrossRef CAS PubMed.
- J. K. Chen and C. J. Chang, Materials, 2014, 7, 805–875 CrossRef CAS PubMed.
- Z. L. Yao, N. Grishkewich and K. C. Tam, Soft Matter, 2013, 9, 5319–5335 RSC.
- C. J. Chang and E. H. Kuo, Thin Solid Films, 2010, 519, 1755–1760 CrossRef CAS PubMed.
- M. Patenaude and T. Hoare, ACS Macro Lett., 2012, 1, 409–413 CrossRef CAS.
- D. Parasuraman and M. J. Serpe, ACS Appl. Mater. Interfaces, 2011, 3, 4714–4721 CAS.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7435 RSC.
- K. T. Oh, H. Yin, E. S. Lee and Y. H. Bae, J. Mater. Chem., 2007, 17, 3987–4001 RSC.
- Y. Chen, N. Ballard, O. D. Coleman, I. J. Hands-Portman and S. A. F. Bon, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1745–1754 CrossRef CAS PubMed.
- S. Nayak and L. A. Lyon, Chem. Mater., 2004, 16, 2623–2627 CrossRef CAS.
- I. Gorelikov, L. M. Field and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 15938–15939 CrossRef CAS PubMed.
- M. Smiglak, J. M. Pringle, X. Lu, L. Han, S. Zhang, H. Gao, D. R. MacFarlaneand and R. D. Rogers, Chem. Commun., 2014, 50, 9228–9250 RSC.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angel, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- C. Yan and T. Mu, Phys. Chem. Chem. Phys., 2014, 16, 5071–5075 RSC.
- M. Watanabe, S. Zhang and K. Dokko, Mater. Horiz., 2015, 2, 168–197 RSC.
- L. C. Tomé, C. Florindo, C. S. R. Freire, L. P. N. Rebelo and I. M. Marrucho, Phys. Chem. Chem. Phys., 2014, 16, 17172–17182 RSC.
- A. Kumar, A. Rani and P. Venkatesu, Phys. Chem. Chem. Phys., 2014, 16, 15806–15810 RSC.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS PubMed.
- C. Guerrero-Sanchez, T. Erdmenger, P. Šereda, D. Wouters and U. S. Schubert, Chem.–Eur. J., 2006, 12, 9036–9045 CrossRef CAS PubMed.
- P. M. Reddy and P. Venkatesu, J. Colloid Interface Sci., 2014, 420, 166–173 CrossRef PubMed.
- C. J. Chang, P. M. Reddy, S. R. Hsieh and H. C. Huang, Soft Matter, 2015, 11, 785–792 RSC.
- N. Winterton, J. Mater. Chem., 2006, 16, 4281–4293 RSC.
- L. Zheng, C. Guo, J. Wang, X. Liang, S. Chen, J. Ma, B. Yang, Y. Jiang and H. Liu, J. Phys. Chem. B, 2007, 111, 1327–1333 CrossRef CAS PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CAS.
- R. Walter, J. Ricka, C. H. Quellet, R. Nuffenger and T. H. Binkert, Macromolecules, 1996, 29, 4019–4028 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- Y. Fang, J. C. Qiang, D. D. Hu, M. Z. Wang and Y. L. Cui, Colloid Polym. Sci., 2001, 279, 14–21 CAS.
- A. Shpigelman, I. Portnaya, O. Ramon and Y. D. Livney, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2307–2318 CrossRef CAS PubMed.
- J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS PubMed.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- P. M. Reddy and P. Venkatesu, J. Phys. Chem. B, 2011, 115, 4752–4757 CrossRef CAS PubMed.
- Z. Wang and P. Wu, RSC Adv., 2012, 2, 7099–7108 RSC.
- R. A. Stile and K. E. Healy, Biomacromolecules, 2001, 2, 185–194 CrossRef CAS PubMed.
- L. M. Mikheeva, N. V. Grinberg, A. Y. Mashkevich, V. Y. Grinberg, L. T. M. Thanh, E. E. Makhaeva and A. R. Khokhlov, Macromolecules, 1997, 30, 2693–2699 CrossRef CAS.
- W. Xue and I. W. Hamley, Polymer, 2002, 43, 3069–3077 CrossRef CAS.
- W. S. Cai, L. H. Gan and K. C. Tam, Colloid Polym. Sci., 2001, 279, 793–799 CAS.
- M. Keerl and W. Richtering, Colloid Polym. Sci., 2007, 285, 471–474 CAS.
- Y. Wang, H. R. Li and S. J. A. Han, J. Phys. Chem. B, 2006, 110, 24646–24651 CrossRef CAS PubMed.
- N. Debeljuh, C. J. Barrow, L. C. Henderson and N. Byrne, Chem. Commun., 2011, 47, 6371–6373 RSC.
- M. Y. Jiang, X. J. Ju, L. Fang, Z. Liu, H. R. Yu, L. Jiang, W. Wang, R. Xie, Q. Chen and L. Y. A. Chu, ACS Appl. Mater. Interfaces, 2014, 6, 19405–19415 CAS.
- K. S. Liao, H. Fu, A. Wan, J. D. Batteas and D. E. Bergbreiter, Langmuir, 2009, 25, 26–28 CrossRef CAS PubMed.
- C. Yan and T. Mu, Phys. Chem. Chem. Phys., 2015, 17, 3241–3249 RSC.
- H. Zein and R. Winter, Phys. Chem. Chem. Phys., 2000, 2, 4545–4551 RSC.
- G. Panick, R. Malessa, R. Winter, G. Rapp, K. J. Frye and C. J. Royer, J. Mol. Biol., 1998, 275, 389–402 CrossRef CAS PubMed.
- Y. Katsumoto, T. Tanaka and Y. Ozaki, J. Phys. Chem. B, 2005, 109, 20690–20692 CrossRef CAS PubMed.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2002, 35, 10172–10177 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18246h |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |