
Graphene oxide/rhodanine redox chemistry and its application in designing high-performance elastomer/graphene composites†
Zhijun Yang,
Wenyi Kuang,
Peijin Weng,
Zhenghai Tang and
Baochun Guo*
Department of Polymer Materials and Engineering, South China University of Technology, Guangzhou, 510640, P. R. China. E-mail: psbcguo@scut.edu.cn; Fax: +86 20 22236688; Tel: +86 20 87113374
First published on 2nd October 2015
Abstract
Reduced graphene oxide (RGO) was prepared by the reduction of graphene oxide (GO) with rhodanine. During the reduction, rhodanine was converted into polyrhodanine via oxidative polymerization initiated by GO, and the RGO was subsequently decorated with polyrhodanine. The reduction process and polymerization have been verified. Rhodanine reduced GO with high efficiency. Additionally, rhodanine is polymerized during the GO reduction process. The wrapping of polyrhodanine onto RGO is also verified. Using this novel modification process, the elastomer/graphene composites were prepared by the in situ interfacial modification of elastomer/GO compounds with rhodanine during the processing. Because of the unique reactivity of polyrhodanine during curing, strong interfaces were observed to form in the resulting elastomer/RGO composites. Furthermore, because of the substantially improved interfacial adhesion, combined with the improved dispersion state, the elastomer/RGO composites exhibited significantly improved mechanical properties compared with the elastomer/GO composites. Regarding the facile process and strikingly high modification efficiency, the present work offers new insight into designing high-performance elastomer/graphene composites by combining interfacial chemistry and curing chemistry.
Introduction
Graphene, which consists of one-atom-thick, two-dimensional layers of carbon, has attracted tremendous attention in recent years because of its unparalleled mechanical, electrical, and thermal properties.1 Currently, various techniques have been explored for the preparation of graphene, including micromechanical cleavage of graphite,2 epitaxial growth,3 chemical vapour deposition4 and solution-based chemical reduction of graphene oxide (GO).5 The latter method has been regarded as the most feasible approach for the large-scale production of graphene and the subsequent processing of graphene-based nanocomposites. The solution-based reduction of GO in the presence of various reducers would result in irreversible aggregation because of the strong van der Waals forces unless external stabilizers were added. Furthermore, even if highly soluble graphene were realized, graphene aggregation could still occur during the removal of the solvents in the subsequent processing of graphene-based composites. Therefore, exploring effective, solvent-free processing technologies, for instance, in situ reduction, for polymer/graphene composites is highly desirable.Recently, considerable efforts have been focused on technologically attractive elastomer/graphene composites. For example, Potts et al. incorporated reduced graphene oxide (RGO) into natural rubber (NR) by latex mixing and studied the processing–morphology–property relationships of the composites in detail.6 However, because of the strong van der Waals interaction forces and the chemically inert surface, difficulties in improving the dispersion quality and interfacial interactions between graphene and the rubber matrix have been critical problems facing rubber/graphene composites. Alternatively, GO, which also possesses good mechanical properties, has been widely used to reinforce rubbers. Benefitting from strengthened interfacial interactions because of its surface oxygenic groups, GO exhibits superior reinforcing efficiency for polar rubbers, such as carboxylated nitrile rubber (XNBR),7 butadiene–styrene–vinyl pyridine rubber (VPR),8 and epoxidized natural rubber (ENR).9 However, a special interfacial design or surface modification was necessary to achieve substantial reinforcement in non-polar rubbers because GO exhibits limited compatibility with these materials. For example, Mao et al. improved the reinforcing efficiency of GO in a SBR matrix by utilizing a small amount of VPR as an interfacial bridge to improve the dispersion quality of GO.10 Additionally, Wu et al. developed a facile in situ reduction method for GO in a rubber matrix to prepare SBR/graphene composites.11 In that work, both the molecular level dispersion of graphene sheets and greatly enhanced mechanical properties were achieved. Despite these results, even if the graphene was well dispersed in the rubber matrix, the lack of intelligent interfacial design between graphene and the rubber matrix aimed at efficient stress transfer will limit the reinforcement provided by graphene in elastomers. However, by combining the in situ reduction of GO and surface modification of the resultant graphene with active substances for subsequent interfacial design may address the issue of both dispersion quality and interfacial strength.
Rhodanine, which is derived from thiazolidine, has been used as a vulcanization accelerator in the rubber industry.12–14 However, rhodanine tends to react rapidly with sulfur molecular and transform into inactive crosslink precursor. Accordingly, curing with rhodanine results in scorching and a low curing rate, which are undesirable in the rubber industry. Thus, use of rhodanine in the rubber industry has slowly decreased. However, this low accelerating effect actually represents an opportunity in the interfacial design if rhodanine or its derivatives are anchored onto the filler that serves as a link between the rhodanine derivatives and the formed rubber chains. Recently, rhodanine has attracted attention because of its unique redox behaviour and potential applications, including as an antibacterial material and an adsorbent for water purification.15–18 Typically, the oxidative polymerization of rhodanine is induced by metal ions, such as ferric and silver ions.17,19 For example, Tang et al. fabricated polyrhodanine-coated cellulose nanocrystals by the in situ polymerization of rhodanine using Fe(III) ions as the initiator.20 Because GO has long been recognized as having strong oxidizing properties,21 we proposed that GO could induce the in situ polymerization of rhodanine and generate modified graphene with an active surface, which may be a promising method for constructing strong interfacial interactions between graphene and the rubber matrix.
In the present work, the redox reaction between GO and rhodanine was confirmed by fully characterizing both the reduction and oxidative products, i.e., RGO and polyrhodanine, and the reduction mechanism was proposed. Based on the redox chemistry between GO and rhodanine, SBR/RGO composites were accordingly prepared by the in situ interfacial modification of elastomer/GO compounds with rhodanine. Significant enhancements in mechanical properties were achieved because of the largely improved interfacial adhesion and dispersion quality, and the formation mechanism of the strong interfacial interactions in the composites was also proposed.
Experimental
Raw materials
SBR latex (Intex 132) with a solid content of 66 wt% and a styrene content of 25 wt% was produced by Polimeri Europa (Italy). Rhodanine (99% pure) was purchased from InnoChem Science & Technology Co. Ltd (Beijing, China). Natural graphite power was purchased from Shanghai Colloidal Co. Ltd (Shanghai, China). All chemicals used for graphite oxidation, including concentrated sulfuric acid, sodium nitrate, potassium permanganate and hydrogen peroxide, were analytical reagents and were used as received. Other rubber additives were of industrial grade and were used as received.Synthesis of reduced graphene oxide (RGO) and thermally treated graphene oxide (TGO)
Graphite oxide (GO) was prepared by oxidizing natural graphite according to a modified Hummers method.22 Reduced graphene oxide (RGO) was synthesized as follows. Graphite oxide was re-dispersed in water via sonication to form GO aqueous solution (2 mg ml−1). Subsequently, excess rhodanine monomer (10-fold excess relative to GO) was added, and the reaction was allowed to proceed for 12 h at 80 °C under vigorous stirring. The resulting black mixture was filtered, and the obtained mixture was then subjected to intense sonication and vigorous stirring in ethanol. The suspension was precipitated by centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, and the supernatant was removed. This process was repeated multiple times to ensure the maximum possible removal of polyrhodanine. Finally, the sediment was freeze-dried for further characterization. A control experiment was carried out to exclude the effect of thermal treatment on GO reduction. The thermally treated GO (TGO) was prepared without rhodanine under the same condition (12 h @ 80 °C).
000 rpm, and the supernatant was removed. This process was repeated multiple times to ensure the maximum possible removal of polyrhodanine. Finally, the sediment was freeze-dried for further characterization. A control experiment was carried out to exclude the effect of thermal treatment on GO reduction. The thermally treated GO (TGO) was prepared without rhodanine under the same condition (12 h @ 80 °C).
Oxidative polymerization of rhodanine in the presence of GO
Polyrhodanine was prepared using a process similar to that used for RGO. Rhodanine (10 wt% relative to GO) was added into the GO suspension (2 mg ml−1), and the mixture was maintained at 80 °C for 12 h under vigorous stirring. The resulting black mixture was filtered, and the obtained mixture was then subjected to intense sonication and vigorous stirring in ethanol. The suspension was precipitated by centrifugation at 14000 rpm, and the supernatant was carefully transferred to a clean beaker. This process was repeated several times. Finally, the polyrhodanine was collected by rotating evaporation and subjected to further characterization.
Preparation of SBR/GO composites
The SBR/GO composites were prepared by latex co-coagulation of SBR latex with a GO suspension. The desired amount of GO aqueous suspension (2 mg ml−1) was added dropwise into the SBR latex, followed by vigorous stirring at room temperature overnight. Subsequently, the mixture was co-coagulated with calcium chloride aqueous solution (1 wt%) and washed with deionized water several times, followed by drying under vacuum at 50 °C for 24 h. The rubber ingredients were mixed with the dried SBR/GO compounds with a two-roll mill and subjected to compression at 160 °C for the optimum curing time, which was determined by a U-CAN UR-2030 vulcameter. The samples are referred to as GOx, where x denotes the GO content (phr) in the composite. The formulations of all the composites are expressed as parts per hundreds of rubber (phr), and the basic recipe is as follows: SBR 100 phr; zinc oxide 5 phr; stearic acid 1 phr; N-isopropyl-N′-phenyl-4-phenylenediamine (4010 NA) 1.5 phr; dibenzothiazoledisulfide (DM) 0.5 phr; N-cyclohexyl-2-benzothiazole (CZ) 1.5 phr; and sulfur 1.5 phr.Preparation of SBR/RGO composites
The SBR/RGO composites were prepared using a method similar to that used for the SBR/GO composites with a small modification: extra rhodanine (33.3 wt% relative to GO) was added into the dried SBR/GO compound by a two-roll mill and subjected to hot-press treatment at 150 °C for 10 minutes prior to the typical protocol. During the hot-press procedure, the rhodanine monomers were believed to migrate and adsorb onto the surface of the GO sheets because of the strong hydrogen-bond interactions, and thus, the in situ reduction of GO and the oxidative polymerization of rhodanine proceeded. The samples are referred to as RGOx, where x denotes the initial GO content (phr) in the composite.Preparation of SBR/rhodanine composites
To evaluate the influence of rhodanine on the vulcanization of rubber, SBR/rhodanine composites with various rhodanine contents were prepared. The rhodanine was mixed with the SBR using a two-roll miller together with the other rubber ingredients. The obtained samples are referred to as Rh-x, where x denotes three times the rhodanine content (phr) in the composite.Measurements
Fourier transform infrared spectra (FTIR) spectra were collected on a Bruker Vertex 70 FTIR spectrometer. Thermal gravimetric analysis (TGA) was performed using a TGA Q500 instrument (USA) under nitrogen purging at a heating rate of 10 °C min−1. X-ray spectroscopy (XPS) analysis was carried out on a Kratos Axis Ultra DLD equipped with a Al Kα radiation source (1486.6 eV). X-ray diffraction (XRD) analysis was performed on an X'pert Pro diffractometer with Cu-Kα radiation (λ = 0.1542 nm). Raman spectra were collected on a LabRAM Aramis Raman spectrometer (HORIBA Jobin Yvon, France), which was equipped with a He–Ne ion laser (532.0 nm) as an excitation source. Ultraviolet-visible (UV-Vis) spectra were obtained using a Scinco S-3150 spectrometer (Korea). For UV-Vis spectroscopy, the samples were dissolved in absolute ethanol. The curing characteristics of the SBR compounds were determined at 160 °C by a U-CAN UR-2030 vulcameter (Taiwan). Dynamic mechanical analysis (DMA) was performed on a TA DMA Q800 instrument under a tensile mode with a dynamic strain of 0.5%. The frequency and heating rate were 5 Hz and 3 °C min−1, respectively. The cryogenically fractured surfaces of the rubber composites were observed using a field emission scanning electron microscope (SEM, Hitachi S-4800, Japan). Before observation, the fracture surfaces were plated with a thin layer of gold. Crosslink density was determined by the equilibrium swelling method based on the Flory–Rehner equation23,24 using toluene as the solvent. Tensile tests were performed on a U-CAN UT-2060 (Taiwan), which was used to measure the tensile strength, modulus and elongation at break according to ISO standard 37-2005.Results and discussion
GO/rhodanine redox chemistry
Herein, rhodanine was employed as an efficient reducing agent for GO. During the redox reaction, GO was reduced to RGO by removing the oxygen-containing groups and recovering the conjugated structure. Additionally, the rhodanine monomers undergo oxidative polymerization and are converted into polyrhodanine. To begin with, the reduction product, RGO, was characterized in detail. As seen in Fig. 1a, the FTIR spectrum of GO shows strong absorption peaks at approximately 3407 cm−1 and 1730 cm−1, which can be assigned to the stretching vibrations of hydroxyl groups and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of –COOH groups, respectively.25 The absorption at 1625 cm−1 can be ascribed to the residual water and the contributions of the skeletal vibrations of un-oxidized graphitic domains.26 Moreover, the characteristic peaks of other oxygen-containing groups, such as carboxy (C–O, 1401 cm−1), epoxy (C–O–C, 1227 cm−1 and 971 cm−1) and alkoxy (C–O, 1057 cm−1) groups, can also be identified in the spectrum of GO.27 As shown in the spectrum of RGO, upon treatment with rhodanine, the intensities of the absorption peaks corresponding to the oxygenic groups mentioned above decrease significantly or disappear completely. In addition, new bands at 1567 and 1485 cm−1, corresponding to CC stretching modes, and the characteristic absorption at approximately 1158 cm−1, which is attributed to the C–C stretching mode, appear.28 This observation strongly supports the reduction of GO by rhodanine. However, it should be noted that there are still some residual oxygenic groups in RGO, which might greatly facilitate the adsorption of the oxidation product of rhodanine. TGA analysis provides further evidence of the reduction of GO. As shown in Fig. 1b, GO is thermally unstable, which is consistent with previous reports in the literature.29 Below 100 °C, 6 wt% weight loss occurs, which can be ascribed to the evaporation of adsorbed water and is characteristic of the intrinsic hydrophilicity of GO. For RGO, less than 2.5% mass loss is observed within this temperature range, which indicates the different surface chemical properties of RGO. The mass loss of GO mainly occurs at approximately 200 °C and can be ascribed to the pyrolysis of labile oxygen-containing groups, such as O–H, CO, and O–CO groups. After the reaction with rhodanine, no obvious mass loss corresponding to the labile oxygen functionalities occurs, which indicates that most of the oxygenic groups have been removed in RGO. Notably, RGO exhibits significant weight loss at approximately 400 °C, which is a typical decomposition temperature for polymers. The weight loss in this range is due to the decomposition of the adsorbed polyrhodanine formed during the reduction of GO.16,30 TGA results showed that the chemical reduction of GO by rhodanine is effective at improving the thermal stability of RGO despite the existence of adsorbed organics (polyrhodanine), which supports the successful conversion of GO into RGO.
O of –COOH groups, respectively.25 The absorption at 1625 cm−1 can be ascribed to the residual water and the contributions of the skeletal vibrations of un-oxidized graphitic domains.26 Moreover, the characteristic peaks of other oxygen-containing groups, such as carboxy (C–O, 1401 cm−1), epoxy (C–O–C, 1227 cm−1 and 971 cm−1) and alkoxy (C–O, 1057 cm−1) groups, can also be identified in the spectrum of GO.27 As shown in the spectrum of RGO, upon treatment with rhodanine, the intensities of the absorption peaks corresponding to the oxygenic groups mentioned above decrease significantly or disappear completely. In addition, new bands at 1567 and 1485 cm−1, corresponding to CC stretching modes, and the characteristic absorption at approximately 1158 cm−1, which is attributed to the C–C stretching mode, appear.28 This observation strongly supports the reduction of GO by rhodanine. However, it should be noted that there are still some residual oxygenic groups in RGO, which might greatly facilitate the adsorption of the oxidation product of rhodanine. TGA analysis provides further evidence of the reduction of GO. As shown in Fig. 1b, GO is thermally unstable, which is consistent with previous reports in the literature.29 Below 100 °C, 6 wt% weight loss occurs, which can be ascribed to the evaporation of adsorbed water and is characteristic of the intrinsic hydrophilicity of GO. For RGO, less than 2.5% mass loss is observed within this temperature range, which indicates the different surface chemical properties of RGO. The mass loss of GO mainly occurs at approximately 200 °C and can be ascribed to the pyrolysis of labile oxygen-containing groups, such as O–H, CO, and O–CO groups. After the reaction with rhodanine, no obvious mass loss corresponding to the labile oxygen functionalities occurs, which indicates that most of the oxygenic groups have been removed in RGO. Notably, RGO exhibits significant weight loss at approximately 400 °C, which is a typical decomposition temperature for polymers. The weight loss in this range is due to the decomposition of the adsorbed polyrhodanine formed during the reduction of GO.16,30 TGA results showed that the chemical reduction of GO by rhodanine is effective at improving the thermal stability of RGO despite the existence of adsorbed organics (polyrhodanine), which supports the successful conversion of GO into RGO.
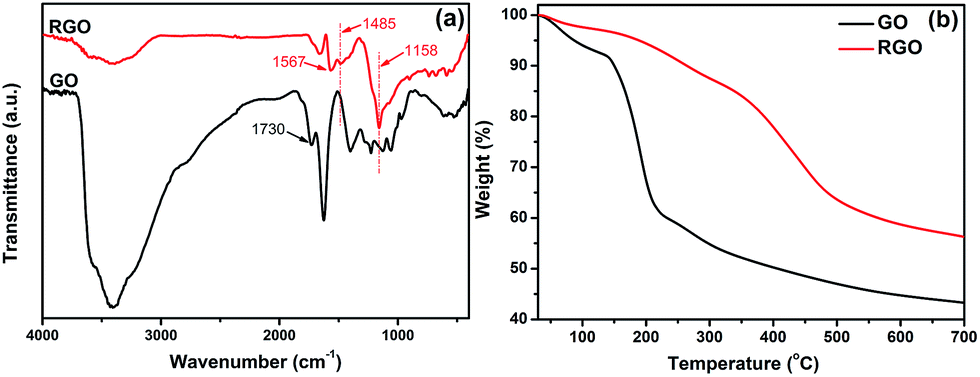 | ||
| Fig. 1 (a) FTIR spectra and (b) TGA curves for GO and RGO. | ||
To further investigate the elemental composition and the reducing effects of rhodanine on GO, XPS measurements were collected. As seen in Fig. 2a, the representative full spectrum of GO shows clear C 1s and O 1s peaks at ca. 284 eV and 532 eV, respectively. However, in addition to the two dominant C 1s and O 1s peaks, three relatively small signals at ca. 163 eV, 227 eV, and 399 eV, corresponding to S 2p, S 2s, and N 1s peaks, respectively,31 are observed in the spectrum of RGO (Fig. 2b). This observation confirms the presence of adsorbed polyrhodanine on the surface of RGO sheets, which is in accordance with the TGA result. Moreover, the C/O atomic ratio calculated from the XPS survey spectrum is ca. 1.7 for GO and is increased to 6.7 after rhodanine reduction, indicating the effective removal of oxygen-containing functionalities. The de-convoluted C 1s XPS spectra of GO and RGO are shown in Fig. 2c and d. The C 1s spectra of GO displays five fitted peak components with binding energies of approximately 283.8, 284.4, 286.4, 287.8, and 289 eV, corresponding to the sp2 carbon, sp3 carbon, C–O, CO, and O–CO groups, respectively.32,33 Upon treatment with rhodanine, the oxidized carbon group content of RGO clearly decreases, as seen in Fig. 2d. These data can be curve-fitted into five peak components with binding energies of approximately 284.4, 285.5, 286.4, 287.8, and 289 eV, which are attributable to the C–C, C–N/C–S, C–O, CO/CN, and O–CO species, respectively.32–35 In order to exclude the effect of thermal treatment on GO reduction, the thermally treated GO (TGO) was also characterized by XPS measurement. As can be seen in Fig. S1,† the representative full spectrum and C 1s spectrum of TGO were similar to those of GO (Fig. 2a and c). According to the calculation of the XPS survey spectrum, the C/O atomic ratio for TGO is less than 2.2, which is very close to the value of GO and is far below the C/O ratio of RGO. Meanwhile, the de-convoluted C 1s XPS spectra of GO displays similar peak components to that of GO which strongly indicates that the oxygenic groups on GO sheets could not be effectively removed by simple heat treatment at 80 °C. However, the reduced oxygen components and dominant single peak corresponding to C–C bonds of RGO suggests the increased aromaticity of RGO sheets and implies the reducing effect of rhodanine on GO. In addition, the presence of C–N, C–S, and CN further indicates the existence of polyrhodanine residues complexed with RGO sheets.
 | ||
| Fig. 2 Full XPS and C 1s spectra of GO (a and c) and RGO (b and d). | ||
The XRD patterns of the crystal structures of GO and RGO are compared in Fig. 3a. The pattern of GO shows a typical (001) diffraction peak at approximately 10.3°, corresponding to a d-spacing of 0.86 nm.36,37 Meanwhile, the diffraction peak at 26.5° (d-spacing of 0.34 nm) for graphite is completely absent,38 which can be ascribed to the presence of oxygenic groups on the graphite sheets. For RGO, the characteristic peak of GO is absent, and a broad diffraction peak appears at approximately 25.8° (d-spacing of 0.35 nm), which is similar to the typical diffraction peak of graphite. The above results further indicate the successful reduction of GO by rhodanine and the restoration of the graphene structure. Raman spectra are useful for characterizing carbonaceous materials, especially graphene. The significant structural changes that occur during the reduction process are reflected in the Raman spectra of GO and RGO, as shown in Fig. 3b. In the Raman spectrum of GO, two dominant peaks, denoted as D band and G band, can be observed. The D band at 1348 cm−1 corresponds to the vibration of sp3 hybridized carbon and defects in disordering structure, while the G band at 1590 cm−1 is related to the vibration of sp2 hybridized carbon in the graphite lattice.37 Compared with GO, the D and G bands of RGO are red shifted to 1330 cm−1 and 1564 cm−1, respectively, possibly because of the restored conjugated structure of graphene. The Raman shifts of the D and G bands for RGO are closer to those of raw graphite, indicating the effective reduction of GO.39 The intensity ratio of the D and G bands (ID/IG), which represents the ratio of disordered and ordered carbon structures, is often adopted to evaluate the defects in graphene and monitor the its functionalization. As shown in Fig. 3b, upon treatment with rhodanine, the ID/IG ratio of GO significantly decreases from 0.95 to 0.26, suggesting that the ordered structure of graphene is recovered after the reduction. In addition, the Raman spectra of graphene show two shoulder peaks at approximately 1495 cm−1 and 1680 cm−1 that can be ascribed to the adsorption of polyrhodanine on the surface of the graphene sheets; similar results have been observed for pyrrole-reduced graphene.40
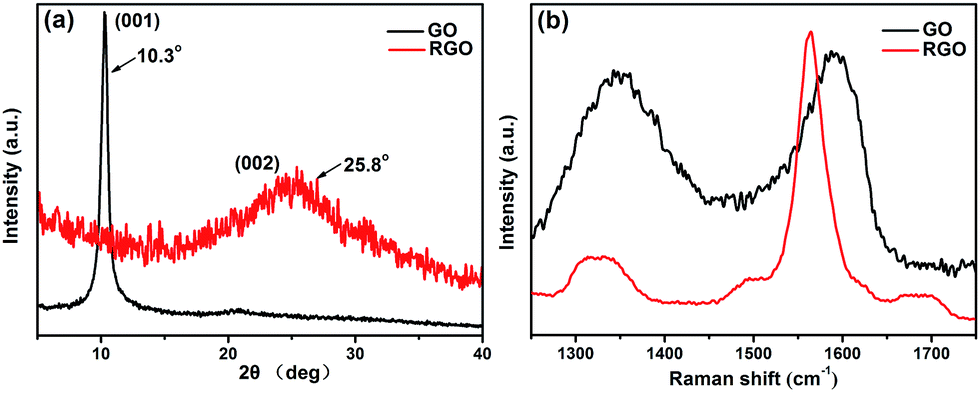 | ||
| Fig. 3 (a) XRD patterns and (b) Raman spectra of GO and RGO. | ||
To further elucidate the redox reaction mechanism between GO and rhodanine, the oxidative products adsorbed on the graphene sheets were extracted and characterized by FTIR and UV-Vis spectra. As seen in Fig. S2,† the extracted products exhibited a purple colour in ethanol, similar to that shown by polyrhodanine polymerized in the presence of metal ions or by electrochemical polymerization.16,18 The simultaneous oxidative polymerization of rhodanine triggered by GO was verified by FTIR and UV-Vis spectroscopy. The FTIR spectra of rhodanine and polyrhodanine are compared in Fig. 4a. It can be seen that although some characteristic adsorption peaks of rhodanine are maintained, some additional adsorption peaks are present in the spectrum of polyrhodanine. The split peaks between 2960 cm−1 and 2850 cm−1 are assigned to the C–H stretching vibration of the methylene group and OC–H structure in the heterocyclic ring.20,41 The peak at approximately 1720 cm−1 is assigned to the stretching vibration of CO. In the spectrum of polyrhodanine, the adsorption peak corresponding to N–H vibration at approximately 3100 cm−1 almost disappears, and new peaks at 1664, 1579, 1285, and 1128 cm−1 are observed. The peak at 1664 cm−1 is attributed to CC stretching vibration, and the band at 1579 cm−1 is assigned to the CN stretching vibration of synthesized polyrhodanine.15,19,42 Moreover, the adsorption bands at 1285 and 1128 cm−1 can be ascribed to C–O stretching vibration.30 Based on the FTIR spectra, the oxidative polymerization of rhodanine induced by GO can be proposed to proceed via the coupling of carbon and nitrogen atoms by forming CC, CN, and C–O bonds. This polymerization is consistent with the results of the electrochemical polymerization of polyrhodanine.16 The UV-Vis spectra also indicate the successful oxidative polymerization of rhodanine (Fig. 4b). For rhodanine monomer, a characteristic absorption peak was observed at 384 nm, which was attributed to its n–π* transition.18,19 In contrast, the extracted product exhibited an adsorption peak at approximately 510 nm, which is correlated with the π–π* transition of the conjugated polyrhodanine backbone.16–19 The above results confirm that the oxidative polymerization of rhodanine accompanies the synchronous reduction of GO. Based on the above characterization of the resulting RGO and polyrhodanine, the reduction mechanism was proposed as shown in Scheme 1.
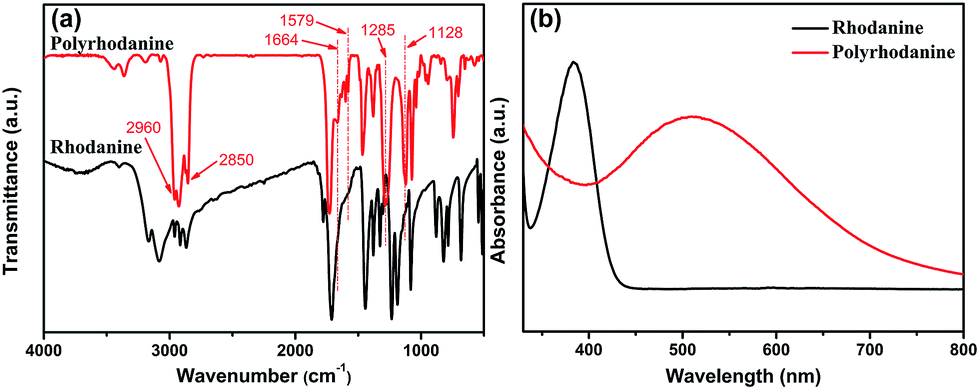 | ||
| Fig. 4 (a) FTIR spectra and (b) UV-Vis spectra of rhodanine and polyrhodanine. | ||
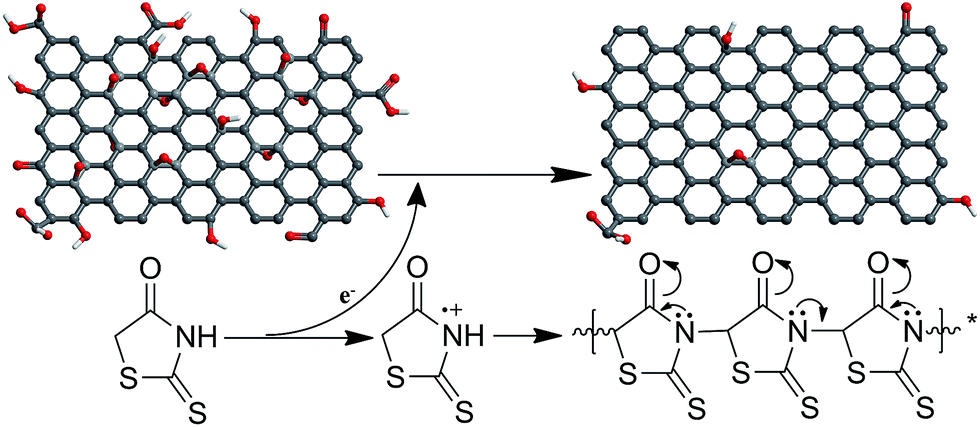 | ||
| Scheme 1 Proposed mechanism of GO reduction by rhodanine. | ||
In the present work, rhodanine was adopted as a reducing agent and surface modifier for GO. During the reduction, GO is partially reduced into graphene, while the rhodanine monomers undergo oxidative polymerization and are converted into polyrhodanine. The oxidation of rhodanine releases electrons that are transferred to GO sheets,16,43 resulting in the reduction of GO. The resulting polyrhodanine, which are possibly rhodanine oligomers, is then anchored onto the surface of the graphene sheets. Because polyrhodanine contains plentiful oxygen and nitrogen groups in its monomeric unit, hydrogen bonding may occur between polyrhodanine and residual oxygen-containing functionalities, such as carboxyl and hydroxyl groups, in the RGO. Moreover, π–π interactions, which were observed in the UV-Vis spectra, also contribute to the intensive adsorption of polyrhodanine onto the surface of the graphene sheets.
Formation of rubber/RGO composites by in situ modification with rhodanine
Previously, we reported that the surface chemistry of the filler is an important factor in determining the composite properties by affecting the dispersion status of the fillers and the interfacial adhesion between the filler and polymer matrix.44 It has been demonstrated that GO can be reduced by inducing the in situ polymerization of rhodanine monomers on the surface of GO sheets. Therefore, SBR/GO and SBR/RGO composites were prepared, and the mechanical properties of the two systems were compared. Representative stress–strain curves for the SBR/GO and SBR/RGO composites with various filler loadings are depicted in Fig. 5a. Explicitly, the ultimate stresses of the composites are significantly improved by the incorporation of both GO and RGO. As expected, SBR/RGO composites exhibit superior performance compared with the SBR/GO composites. The tensile modulus values (at 200% strain) for all the composites are shown in Fig. S3.† The modulus for the SBR/RGO composite is substantially increased (by at least 100%) compared with that of the SBR/GO composite with same filler content. In particular, compared with neat SBR, the modulus of GO7 is increased by 2.8-fold, while that for RGO7 is drastically increased by 7.7-fold. Such enhancements are much more prominent than those reported previously. For example, Wu et al. incorporated chemically reduced graphene into SBR by a modified latex compounding method and in situ reduction of GO.11 The modulus (200% strain) of the as-prepared SBR composite with 7-phr graphene was less than 8 MPa, which was far inferior to that achieved in the present work (12.9 MPa). The significant improvement in the tensile modulus in this work can be primarily ascribed to the strong interfacial interactions resulting from the introduction of polyrhodanine onto the surface of the graphene sheets, which acted as an interfacial bridge between the graphene sheets and the rubber matrix.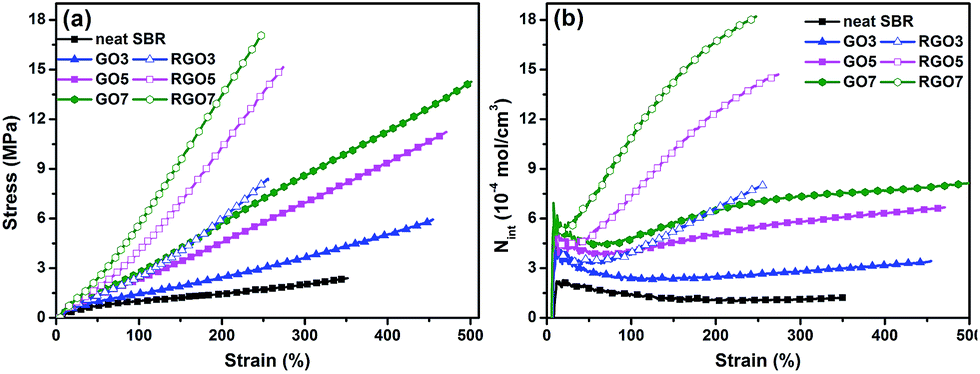 | ||
| Fig. 5 (a) Typical stress–strain curves and (b) dependence of interfacial interactions on the strain for SBR/GO and SBR/RGO composites. | ||
To quantify the interfacial interactions between the filler and the rubber matrix in both systems, a previously proposed equation based on the theory of rubber elasticity and reinforcement was employed:37,45–47
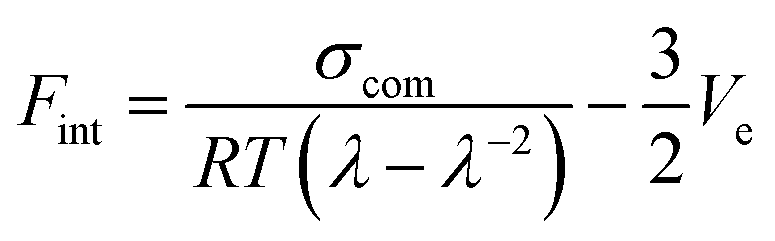
Because it contains abundant oxygenic groups, GO exhibits hydrophilic characteristics that result in poor compatibility with non-polar hydrophobic rubbers. Upon hot-press treatment with rhodanine, GO is partially reduced into graphene. In addition, the resultant polyrhodanine adsorbed onto the graphene surface and triggered in situ interfacial interaction during the subsequent vulcanization. These behaviours are believed to contribute to improving the compatibility between graphene and the SBR matrix. FESEM was utilized to investigate the morphology and evaluate the interfacial adhesion of the SBR composites. The SEM images of cryogenically fractured surfaces of SBR/GO and SBR/RGO composites are compared in Fig. 6. As shown in Fig. 6a, some wrinkled GO protrude from the rubber matrix, indicating weak interfacial interactions between GO and SBR. As the GO loading increases (Fig. 6b and c), more GO sheets are observed to be pulled out of the SBR matrix, and apparent aggregations of GO are also visible. The FESEM images for SBR/RGO composites are shown in Fig. 6d–f. It can be clearly seen that the cryogenically fractured surfaces of SBR/RGO samples exhibit distinctly different morphologies. In these samples, the graphene sheets are uniformly dispersed in the matrix without apparent aggregation. No obvious extraction of GO can be seen, and the interface between the graphene sheets and the rubber matrix are blurry because the graphene is embedded in the SBR matrix. These observations demonstrate that the combination of GO reduction by rhodanine and oxidative polymerization of rhodanine by GO can effectively promote the dispersion of GO and the adhesion between the graphene sheets and the SBR matrix.
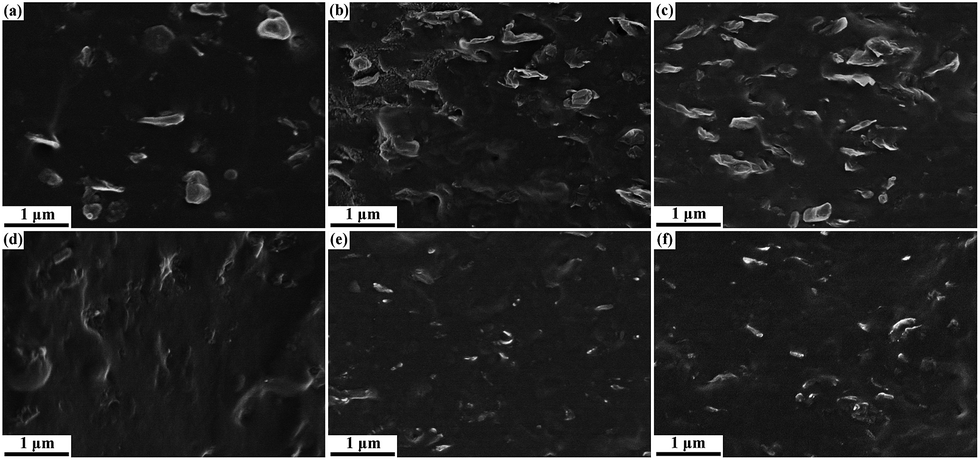 | ||
| Fig. 6 FESEM images of (a) GO1, (b) GO5, (c) GO7, (d) RGO1, (e) RGO5, and (f) RGO7 composites. | ||
To determine the mechanism underlying the significant improvement in the interfacial interactions in SBR/RGO composites, the vulcanization characteristics of SBR/rhodanine, SBR/GO and SBR/RGO compounds were studied, as depicted in Fig. S5 and S6.† Generally, increasing the accelerator should result in simultaneous decreases in both the scorch time (Tc10) and the optimum curing time (Tc90).48 When a small amount of rhodanine is added, both Tc10 and Tc90 are slightly decreased compared with the neat SBR. However, it is clear that Tc10 consistently decreased and that Tc90 consistently increased as the rhodanine loading increased (Fig. S5(a)†). Similar to other accelerators, such as mercaptobenzothiazole (MBT),49,50 rhodanine, which contains –(NH)–(CS)– group, is capable of undergoing tautomerization to form the –NC–SH structure, as shown in Fig. S6.† This process is responsible for the accelerating effect of rhodanine.51–53 However, the existence of carbonyl groups near the –NC–SH groups decreases the nucleophilicity of the nitrogen atom, which is essential for chelation with the zinc ion and the efficient formation of crosslinks, as shown in Fig. S6.† 50 Therefore, rhodanine tends to react with sulfur molecules more rapidly than the adopted accelerator (DM and CZ), and inactive crosslink precursors (monosulfide linkage structures) are formed between rhodanine and the rubber chains. This unique curing behaviour leads to consistently decreasing crosslink density as increasing amounts of rhodanine are added (Fig. S7†).
As the RGO content increases, the evolution of the curing profile is similar to that of SBR/rhodanine compounds. However, compared with SBR/rhodanine compounds, the curing time and scorching time of the SBR/RGO composites are less sensitive to the RGO content. In addition, the long marching behaviour exhibited by SBR/rhodanine compounds is almost absent in SBR/RGO composites. These observations indicate that the SBR/RGO composites experienced an accelerating mechanism similar to that of rhodanine, although the “adverse effects” are substantially lessened. During GO reduction, the rhodanine monomers undergo polymerization to form polyrhodanine. The accelerating effect of rhodanine is preserved in the polyrhodanine because of the presence of the terminal NC–SH moiety,16 which is responsible for the interfacial crosslinking. However, it should be noted that even though RGO7 contains a higher rhodanine content, the polymerization significantly reduces the available content of accelerating rhodanine. Therefore, the adverse effect on crosslinking is highly inhibited.
The curing behaviours of RH-7, GO7 and RGO7 are selectively compared in Fig. 7a. Specifically, the equilibrium torque of RGO7 is higher than that of GO7. At the same filler content, the crosslink density of the SBR/RGO sample is clearly higher than that of the SBR/GO sample (Fig. 7b). Therefore, based on the above discussion, the interfacial crosslinking occurs in SBR/RGO composites, leading to increased overall crosslink density and modulus value. The consistent and rapid increase in the crosslink density observed as the amount of RGO increases provides supporting evidence for the increasing interfacial crosslinking along the increase in RGO content (Fig. 7b).
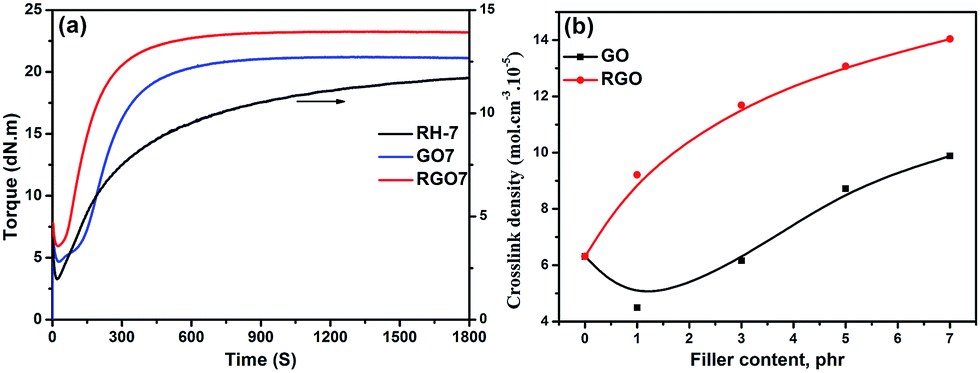 | ||
| Fig. 7 (a) The curing behaviours of RH-7, GO7 and RGO7 and (b) crosslinking density for SBR/GO and SBR/RGO composites. | ||
The schematic illustration of the formation mechanism of the strong interfacial interactions in SBR/RGO composites was proposed based on the above results (Scheme 2). In this structural model, the polyrhodanine molecules adhere to the surface of the graphene sheets through hydrogen bonds and π–π interactions, as shown above. The numerous exposed ends of polyrhodanine chains exert a special accelerating effect on the curing, leading to interfacial crosslinking between graphene sheets and the rubber matrix.
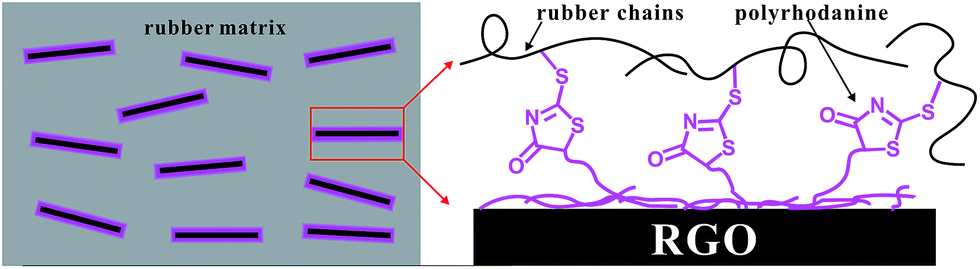 | ||
| Scheme 2 Schematic illustration of the proposed interfacial crosslink mechanism in SBR/RGO composites. | ||
Conclusions
By combining the GO reduction and the oxidative polymerization of rhodanine, samples of RGO with active surfaces were successfully synthesized for the first time using rhodanine as the reducing agent. Interfacial crosslinking between the polyrhodanine-coated graphene sheets and the rubber chains was generated in situ during the vulcanization of SBR/GO composites in the presence of rhodanine. Accordingly, significant enhancement of the modulus of SBR/RGO composites was achieved through the formation of the unique interfacial crosslink structure and improved dispersion. With the incorporation of 7 phr of RGO, the modulus values (at 200% strain) were increased by 7.7-fold compared with that of the neat SBR. The present study revealed the critical role of interfacial adhesion in determining the performance of rubber/graphene composites and provided a novel and facile solution for interfacial design for high performance elastomeric composites.Acknowledgements
We thank the National Basic Research Program of China (2015CB654703), NSFC (No. 51222301, 51473050 and 51333003) and the Fundamental Research Funds for the Central Universities (No. 2014ZG0001).Notes and references
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- K. Novoselov, D. Jiang, F. Schedin, T. Booth, V. Khotkevich, S. Morozov and A. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS PubMed.
- S. W. Poon, W. Chen, E. S. Tok and A. T. S. Wee, Appl. Phys. Lett., 2008, 92, 104102 CrossRef PubMed.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2008, 9, 30 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- J. R. Potts, O. Shankar, L. Du and R. S. Ruoff, Macromolecules, 2012, 45, 6045 CrossRef CAS.
- X. Liu, D. Sun, L. Wang and B. Guo, Ind. Eng. Chem. Res., 2013, 52, 14592 CrossRef CAS.
- Z. Tang, X. Wu, B. Guo, L. Zhang and D. Jia, J. Mater. Chem., 2012, 22, 7492 RSC.
- X. She, C. He, Z. Peng and L. Kong, Polymer, 2014, 55, 6803 CrossRef CAS PubMed.
- Y. Mao, S. Wen, Y. Chen, F. Zhang, P. Panine, T. W. Chan, L. Zhang, Y. Liang and L. Liu, Sci. Rep., 2013, 3, 2508 Search PubMed.
- W. Xing, M. Tang, J. Wu, G. Huang, H. Li, Z. Lei, X. Fu and H. Li, Compos. Sci. Technol., 2014, 99, 67 CrossRef CAS PubMed.
- K. V'yunov, A. Ginak and E. Sochilin, J. Appl. Spectrosc., 1972, 16, 769 CrossRef.
- J. D. Amico, US Pat., 2,721,869, 1955.
- S. Seraj, B. Mirzayi and A. Nematollahzadeh, Adv. Powder Technol., 2014, 25, 1520 CrossRef CAS PubMed.
- J. Song, H. Kim, Y. Jang and J. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 11563 CAS.
- G. Kardaş and R. Solmaz, Appl. Surf. Sci., 2007, 253, 3402 CrossRef PubMed.
- H. Kong, J. Song and J. Jang, Chem. Commun., 2010, 46, 6735 RSC.
- J. Song, H. Song, H. Kong, J.-Y. Hong and J. Jang, J. Mater. Chem., 2011, 21, 19317 RSC.
- H. Kong, J. Song and J. Jang, Macromol. Rapid Commun., 2009, 30, 1350 CrossRef CAS PubMed.
- J. Tang, Y. Song, R. M. Berry and K. C. Tam, RSC Adv., 2014, 4, 60249 RSC.
- H. P. Boehm, A. Clauss, G. Fischer and U. Hofmann, Fifth Conference on Carbon, Pergamon Press, 1962, p. 73 Search PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- P. J. Flory and J. Rehner Jr, J. Chem. Phys., 1943, 11, 521 CrossRef CAS PubMed.
- P. J. Flory, J. Chem. Phys., 1950, 18, 108 CrossRef CAS PubMed.
- Z. Lin, G. Waller, Y. Liu, M. Liu and C.-P. Wong, Adv. Energy Mater., 2012, 2, 884 CrossRef CAS PubMed.
- H.-L. Guo, X.-F. Wang, Q.-Y. Qian, F.-B. Wang and X.-H. Xia, ACS Nano, 2009, 3, 2653 CrossRef CAS PubMed.
- G. I. Titelman, V. Gelman, S. Bron, R. L. Khalfin, Y. Cohen and H. Bianco-Peled, Carbon, 2005, 43, 641 CrossRef CAS PubMed.
- J. Yin, X. Wang, R. Chang and X. Zhao, Soft Matter, 2012, 8, 294 RSC.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514 CrossRef CAS.
- S. Ozkan, H. I. Unal, E. Yılmaz and Z. Suludere, J. Appl. Polym. Sci., 2015, 132, 41554 Search PubMed.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS PubMed.
- L. Q. Xu, W. J. Yang, K. G. Neoh, E.-T. Kang and G. D. Fu, Macromolecules, 2010, 43, 8336 CrossRef CAS.
- I. Kaminska, W. Qi, A. Barras, J. Sobczak, J. Niedziolka-Jonsson, P. Woisel, J. Lyskawa, W. Laure, M. Opallo and M. Li, Chem.–Eur. J., 2013, 19, 8673 CrossRef CAS PubMed.
- H. Bai, Y. Xu, L. Zhao, C. Li and G. Shi, Chem. Commun., 2009, 13, 1667 RSC.
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291 CrossRef CAS PubMed.
- J. Jiang and X. Du, Nanoscale, 2014, 6, 11303 RSC.
- X. Liu, W. Kuang and B. Guo, Polymer, 2015, 56, 553 CrossRef CAS PubMed.
- H. Xiang, K. Zhang, G. Ji, J. Y. Lee, C. Zou, X. Chen and J. Wu, Carbon, 2011, 49, 1787 CrossRef CAS PubMed.
- Z. Tang, L. Zhang, C. Zeng, T. Lin and B. Guo, Soft Matter, 2012, 8, 9214 RSC.
- C. A. Amarnath, C. E. Hong, N. H. Kim, B.-C. Ku, T. Kuila and J. H. Lee, Carbon, 2011, 49, 3497 CrossRef CAS PubMed.
- J. Tang, Y. Song, S. Tanvir, W. A. Anderson, R. M. Berry and K. C. Tam, ACS Sustainable Chem. Eng., 2015, 3, 1801 CrossRef CAS.
- J. Song, H. Oh, H. Kong and J. Jang, J. Hazard. Mater., 2011, 187, 311 CrossRef CAS PubMed.
- J. Song, H. Kong and J. Jang, J. Colloid Interface Sci., 2011, 359, 505 CrossRef CAS PubMed.
- Z. Tang, L. Zhang, W. Feng, B. Guo, F. Liu and D. Jia, Macromolecules, 2014, 47, 8663 CrossRef CAS.
- Y. Lei, Z. Tang, B. Guo, L. Zhu and D. Jia, eXPRESS Polym. Lett., 2010, 4, 692 CrossRef CAS.
- Y. Lei, Z. Tang, L. Zhu, B. Guo and D. Jia, Polymer, 2011, 52, 1337 CrossRef CAS PubMed.
- Y. Lei, Z. Tang, L. Zhu, B. Guo and D. Jia, J. Appl. Polym. Sci., 2012, 123, 1252 CrossRef CAS PubMed.
- W. Siwu, L. Tengfei and G. Baochun, Nanotechnology, 2013, 24, 465708 CrossRef PubMed.
- H. Koch, J. Chem. Soc., 1949, 401 RSC.
- J. E. Mark, B. Erman and M. Roland, The science and technology of rubber, Academic press, 2013 Search PubMed.
- R. Layer, US Pat., 3,763,089, 1973.
- V. Enchev, I. Petkov and S. Chorbadjiev, Struct. Chem., 1994, 5, 225 CrossRef CAS.
- D. Tahmassebi, J. Mol. Struct.: THEOCHEM, 2003, 638, 11 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs of rhodanine and polyrhodanine dispersions, tensile modulus of SBR/GO and SBR/RGO composites, tanδ curves of SBR/GO and SBR/RGO composites, vulcanization curves and curing data; schemes of potential accelerating mechanism, crosslink density for SBR/rhodanine compounds. See DOI: 10.1039/c5ra18098h |
| This journal is © The Royal Society of Chemistry 2015 |