
Highly π-extended tetrathiafulvalene analogues derived from pentacene-5,7,12,14-tetraone†
Abstract
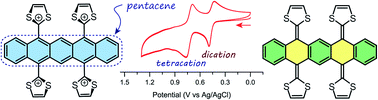
A new class of π-extended tetrathiafulvalene analogue, which is capable of reversibly revealing and concealing a central pentacene segment under redox control, was synthesized by stepwise olefination reactions on pentacene-5,7,12,14-tetraone. The structural, electronic, and redox properties were investigated by NMR, UV-Vis absorption, electrochemical analyses in conjunction with density functional theory (DFT) calculations.
 Please wait while we load your content...
Please wait while we load your content...

