DOI:
10.1039/C5RA17844D
(Paper)
RSC Adv., 2015,
5, 92456-92472
A family of mixed-ligand oxidovanadium(V) complexes with aroylhydrazone ligands: a combined experimental and computational study on the electronic effects of para substituents of hydrazone ligands on the electronic properties, DNA binding and nuclease activities†
Received
2nd September 2015
, Accepted 20th October 2015
First published on 20th October 2015
Abstract
A new series of mixed-ligand oxidovanadium(V) complexes [VVO(HL1–4)(hq)] (1–4) have been synthesized using 2-hydroxybenzoylhydrazones of 2-hydroxyacetophenone and its 5-substituted derivatives (H3L1–4) along with 8-hydroxyquinoline (Hhq) as co-ligand. The complexes were characterized by elemental analyses, magnetic susceptibility measurements and various spectroscopic methods. Their electrochemical behaviour is also reported. X-ray crystallographic investigations of 1–4 show the presence of distorted octahedral geometries with O4N2 coordination environments for each of the four complexes. λmax values for the ligand-to-metal-charge-transfer (LMCT) transition, E½ values and the chemical shift parameters (δ) for the 51V NMR spectra of the complexes exhibit a linear relationship with the Hammett constant (σ) of the substituents. DFT methods were used to predict the bond lengths, bond angles, λmax values for electronic transitions and δ values of 51V NMR spectra, all of which are found to be in good agreement with experimental results. The stability of the complexes was also examined. All the complexes exhibit DNA binding activity with CT-DNA either by minor groove binding mode (for 1 and 4) or by partial intercalation mode (for 2 and 3). The complexes were also tested for DNA nuclease activity with pUC19 plasmid DNA and were found to produce both nicked coiled and linear forms. The DNA binding and nuclease activities of the complexes follow the order: 3 > 2 > 1 > 4, which is also the hydrazone ligands' basicity order, suggesting that the binding and cleavage efficiencies are proportional to the electron density on the vanadium centre. The results of DNA binding experiments are further supported by molecular docking studies.
1. Introduction
It can be argued that the best way of fine tuning the basic strength of a functional group present in an aryl ring is by introducing a suitable substituent at an appropriate position. To realise only the electronic effect (i.e., without any steric effect) of the substituents, the appropriate position is para, with respect to the concerned functional group. Over the past few years, we have been working in this area with a view to establish the effect of substituents in the coordinated ligands on the electronic properties of vanadium in its complexes containing VO2+, VO3+ and V2O34+ motifs.1 The ability of vanadium to exist in different motifs and their facile interconversion at ambient conditions in the presence of suitable ligand environments creates an attractive area of contemporary research for inorganic chemists. On the other hand, vanadium is also a point of increasing interest to biochemists because of its numerous biochemical activities2 viz., the stimulatory effect on the growth of algae and plants,3 the inhibitory action on Na, K-ATPase,4 its presence at the active site of certain enzymes, e.g., haloperoxidases in sea algae and lichens,5 nitrogen fixation,6 insulin mimicking,7–11 tumour growth inhibition and prophylaxis against carcinogenesis12 deserve mentioning. Vanadium complexes also have the ability to suppress the growth of existing tumours by inhibiting tumour cell proliferation, including apoptosis and limiting the invasion and metastatic potential of neoplastic cells.13,14
Hydrazone ligands derived from the condensation of aliphatic/aromatic acid hydrazides with aliphatic/aromatic carbonyl compounds are versatile ligands and have wide applications in the field of analytical15 and medicinal16 chemistry. Hydrazone moieties are important pharmaceutical cores of several anti-cancer, anti-inflammatory and anti-platelet drugs.17 Recently, a report on vanadium hydrazone complexes with potential biological properties has appeared.18
On the other hand, 8-hydroxyquinoline (Hhq), a quinoline derivative originating in plants as well as from synthesis, has been used as a fungicide in agriculture and a preservative in the textile, wood and paper industries.19 Hhq is widely used for analytical and separation purposes as well as for metal chelation due to its potent coordinating ability and good metal recognition properties.20 Of all the hydroxyquinoline derivatives, Hhq is the most interesting because of its multi-functional properties, such as diverse bioactivities and therapeutic potentials.21
Taking this background into account, the synthesis and characterisation of the oxidovanadium(V) complexes with the biologically relevant hydrazone and Hhq ligands have been undertaken to study their chemical properties as well as their DNA binding and cleavage activities. However, such types of mixed-ligand complexes are few in number.1e,22 Moreover, the variation of the basic properties of hydrazone ligands through substitution is also important in designing the potential complexes with better DNA binding and cleaving properties.23 On this context, a variation of substituents in the acetophenone ring of the hydrazone ligand with different electronic properties particularly at the para position with respect to a specific functional group (as it is the most suitable position to realise only the electronic effect of substituent without any steric influence) can finely tune the basic strength of the hydrazone ligand which may have significant effects on the electronic properties of vanadium as well as on the biological properties of resulting complexes. However, the analogous mixed-ligand hydrazone complexes, especially a systematic study concerning their pharmacological activity along with their chemical property with the variation of substituent in the ligand moiety is hitherto unknown and needs to be explored. For this reason, four 2-hydroxybenzoylhydrazone ligands of the 2-hydroxyacetophenone family with varying substituents at the para position with respect to the OH group in the acetophenone ring have been chosen in this study (Chart 1).
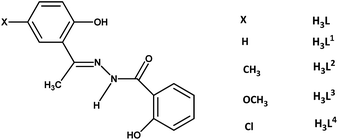 |
| | Chart 1 | |
2. Experimental section
2.1. Materials and methods
2-Hydroxyacetophenone, 2-hydroxy-5-methylacetophenone, 2-hydroxy-5-methoxyacetophenone and 5-chloro-2-hydroxyacetophenone were obtained from Aldrich. 2-Hydroxybenzoylhydrazine was purchased from E. Merck. Acetylacetone, vanadyl sulphate pentahydrate were obtained from Loba chemical company (India). [VIVO(acac)2] was prepared by the reported method.24 All other reagents were of A. R. grade, obtained from commercial sources and used without further purification. Calf thymus (CT) DNA was purchased from Sigma Aldrich and pUC19 DNA was procured from New England Biolabs.
Elemental analyses were performed on a Perkin-Elmer 2400 CHNS/O analyzer. IR spectra (as KBr pellets) were recorded on a Shimadzu FTIR spectrophotometer, IR Affinity-1. Magnetic susceptibilities were measured at 298 K using a PAR model 155 vibrating sample magnetometer fitted with Walker Scientific L75 FBAL magnet using Hg[Co(SCN)4] as the calibrant. The ESI-MS in positive ion mode were measured on a Micromass Qtof YA 263 mass spectrometer. Conductivity measurements of all the complexes were performed at 298 K in CH3CN solution with the Systronics digital conductivity meter. The 1H NMR spectra of complexes were recorded in CDCl3 and of ligands in DMSO-d6 on a Bruker AM 300L either 400 MHz or 500 MHz superconducting FT NMR spectrophotometer and the 51V NMR spectra were recorded on a Bruker 500 MHz spectrophotometer at 293 K in CDCl3 and DMSO-d6 solution. The 51V NMR spectra were referenced to VOCl3 (0 ppm) as an external reference. Electrochemical measurements were performed at 298 K in CH2Cl2 solution for ca. 1 × 10−3 mol dm−3 using tetrabutyl-ammonium perchlorate as the supporting electrolyte under a dry N2 atmosphere on a BASi Epsilon-EC electroanalytical instrument. The BASi platinum working electrode, platinum auxiliary electrode and saturated calomel electrode (SCE) as reference electrode were used for the measurements. The redox potential data are referenced to a ferrocenium/ferrocene, Fc+/Fc couple. UV-vis spectra were recorded on a Shimadzu UV-1700 spectrophotometer. Emission intensity measurements were carried out using an RF-5301PC Shimadzu spectroflourimeter. Circular dichroism (CD) spectra were obtained with a Jasco J-1500 spectropolarimeter. Agarose gel images were captured by the UVP GelDoc-It 310 gel documentation system.
2.2. Synthesis of the ligands (H3L1–4)
Hydrazone ligands, H3L1–4, were synthesised in good yield by refluxing 2-hydroxybenzoylhydrazide with the respective 2-hydroxyacetophenone in equimolar ratio in methanol. The resulting white compounds were filtered, washed with methanol and dried over fused CaCl2. Elemental analysis, 1H NMR, IR and mass spectral data confirmed the identity of all four ligands. The analytical data of H3L1 ligand are not described here as this ligand has been recently reported25 in the literature.
2.2.1. H3L2. Yield: 0.20 g (71%). Melting point: 292 °C. Anal. calc. for C16H16N2O3: C, 67.59; H, 5.67; N, 9.85. Found: C, 67.42; H, 5.50; N, 9.74. IR (KBr, νmax/cm−1): 3452 (br, O–Hphenolic), 3021 (N–H), 1627 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1615 (CNazomethine), 1386 (C–Ophenolic), 1291 (C–Oenolic), 922 (N–N). UV-Vis (CH2Cl2) (ε/dm3 mol−1 cm−1): 345 (8450), 288 (9100). 1H NMR (400 MHz, DMSO-d6, 298 K, δ/ppm): 7.12 (d, 1H, H-3, J = 8.0), 6.82 (d, 2H, H-4, H-13, J = 8.0), 7.46 (d, 2H, H-5, H-12, J = 8.4), 6.99–7.05 (m, 2H, H-6, H-11), 7.98 (dd, 1H, H-14, J = 7.2, 0.8), 2.41 (s, 3H, H-15), 2.27 (s, 3H, H(4-CH3)), 12.91 (s, 1H, H(1-OH)), 11.54 (s, 1H, H(10-OH)). ESI-MS (positive) in CH3CN: 285 (M + H+), 307 (M + Na+).
O), 1615 (CNazomethine), 1386 (C–Ophenolic), 1291 (C–Oenolic), 922 (N–N). UV-Vis (CH2Cl2) (ε/dm3 mol−1 cm−1): 345 (8450), 288 (9100). 1H NMR (400 MHz, DMSO-d6, 298 K, δ/ppm): 7.12 (d, 1H, H-3, J = 8.0), 6.82 (d, 2H, H-4, H-13, J = 8.0), 7.46 (d, 2H, H-5, H-12, J = 8.4), 6.99–7.05 (m, 2H, H-6, H-11), 7.98 (dd, 1H, H-14, J = 7.2, 0.8), 2.41 (s, 3H, H-15), 2.27 (s, 3H, H(4-CH3)), 12.91 (s, 1H, H(1-OH)), 11.54 (s, 1H, H(10-OH)). ESI-MS (positive) in CH3CN: 285 (M + H+), 307 (M + Na+).
2.2.2. H3L3. Yield: 0.23 g (77%). Melting point: 256 °C. Anal. calc. for C16H16N2O4: C, 63.99; H, 5.37; N, 9.33. Found: C, 63.84; H, 5.22; N, 9.22. IR (KBr, νmax/cm−1): 3342 (O–Hphenolic), 2940 (N–H), 1628 (CO), 1616 (CNazomethine), 1387 (C–Ophenolic), 1287 (C–Oenolic), 922 (N–N). UV-Vis (CH2Cl2) (ε/dm3 mol−1 cm−1): 357 (9020), 306 (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 290). 1H NMR (400 MHz, DMSO-d6, 298 K, δ/ppm): 7.13 (d, 1H, H-3, J = 2.8), 6.95 (dd, 2H, H-4, H-13, J = 8.8, 2.8), 6.86 (d, 1H, H-5, J = 8.8), 6.99–7.05 (m, 2H, H-6, H-11), 7.46 (dt, 1H, H-12, J = 7.2, 0.8), 7.97 (dd, 1H, H-14, J = 7.6, 1.2), 2.41 (s, 3H, H-15), 3.74 (s, 3H, H(4-OCH3)), 12.63 (s, 1H, H(1-OH)), 11.55 (s, 1H, H(10-OH)), 11.90 (s, 1H, N–H). ESI-MS (positive) in CH3CN: 301 (M + H+), 323 (M + Na+).
290). 1H NMR (400 MHz, DMSO-d6, 298 K, δ/ppm): 7.13 (d, 1H, H-3, J = 2.8), 6.95 (dd, 2H, H-4, H-13, J = 8.8, 2.8), 6.86 (d, 1H, H-5, J = 8.8), 6.99–7.05 (m, 2H, H-6, H-11), 7.46 (dt, 1H, H-12, J = 7.2, 0.8), 7.97 (dd, 1H, H-14, J = 7.6, 1.2), 2.41 (s, 3H, H-15), 3.74 (s, 3H, H(4-OCH3)), 12.63 (s, 1H, H(1-OH)), 11.55 (s, 1H, H(10-OH)), 11.90 (s, 1H, N–H). ESI-MS (positive) in CH3CN: 301 (M + H+), 323 (M + Na+).
2.2.3. H3L4. Yield: 0.24 g (83%). Melting point: 310 °C. Anal. calc. for C15H13ClN2O3: C, 59.12; H, 4.30; N, 9.19. Found: C, 58.90; H, 4.12; N, 9.09. IR (KBr, νmax/cm−1): 3266 (O–Hphenolic), 3083 (N–H), 1632 (CO), 1613 (CNazomethine), 1284 (C–Ophenolic), 1236 (C–Oenolic), 914 (N–N). UV-Vis (CH2Cl2) (ε/dm3 mol−1 cm−1): 342 (8020), 281 (24000). 1H NMR (400 MHz, DMSO-d6, 298 K, δ/ppm): 7.66 (d, 1H, H-3, J = 0.8), 7.04 (t, 2H, H-4, H-13, J = 7.6), 7.34 (d, 1H, H-5, J = 8.4), 6.95–7.00 (m, 2H, H-6, H-11), 7.46 (t, 1H, H-12, J = 7.2), 7.97 (d, 1H, H-14, J = 7.6), 2.42 (s, 3H, H-15), 13.23 (s, 1H, H(1-OH)), 11.60 (s, 1H, H(10-OH)), 11.88 (s, 1H, N–H). ESI-MS (positive) in CH3CN: 304 (M).
2.3. Synthesis of complexes [VVO(HL)(hq)] (1–4)
2.3.1. [VVO(HL1)(hq)] (1). To a methanolic solution (20 mL) of [VIVO(acac)2] (0.265 g, 1 mmol) was added warm methanolic solution (20 mL) of H3L1 (0.270 g, 1 mmol) dropwise with stirring. The reaction mixture was then heated under reflux for 2 h. After cooling the reaction mixture to room temperature, a methanolic solution (10 mL) of Hhq (0.145 g, 1 mmol) was added dropwise with continuous stirring. Immediately the brown solution turned violet. The shiny black microcrystalline product obtained after stirring the reaction mixture for 1 h at room temperature, was filtered, washed with methanol and dried over silica gel. Yield: 0.45 g (94%). Anal. calc. for C24H18N3O5V: C, 60.13; H, 3.78; N, 8.77. Found: C, 60.02; H, 3.88; N, 8.71. IR (KBr, νmax/cm−1): 3438 (br, O–Hphenolic), 1620, 1588 (CNazomethine), 1367 (C–Ophenolic), 1247 (C–Oenolic), 1027 (N–N), 974 (VO). λmax (CH2Cl2)/nm (ε/dm3 mol−1 cm−1): 544 (4860), 332 (15000). 1H NMR (500 MHz, CDCl3, 298 K, δ/ppm): 7.86 (d, 1H, H-3, J = 8.5), 7.05 (t, 1H, H-4, J = 8.5), 7.67 (t, 1H, H-5, J = 8.5), 6.93 (d, 2H, H-6, H-17, J = 8.5), 7.24–7.30 (m, 3H, H-11, H-18, H-19), 7.48–7.52 (m, 3H, H-12, H-14, H-22), 6.69 (t, 1H, H-13, J = 8.5), 2.97 (s, 3H, 3 × H-15), 8.15 (d, 1H, H-21, J = 8.0), 7.92 (d, 1H, H-23, J = 4.5), 11.33 (s, 1H, O–Hphenolic). 51V NMR (500 MHz, 293 K, δ/ppm): −483.4 (CDCl3), −481.1 (DMSO-d6). ESI-MS (positive) in CH3CN: 480 (M + H+), 502 (M + Na+), 518 (M + K+).
2.3.2. [VVO(HL2)(hq)] (2). This compound was prepared following an identical method as described for compound 1 using the ligand H3L2 instead of H3L1. Yield: 0.44 g (89%). Anal. calc. for C25H20N3O5V: C, 60.86; H, 4.09; N, 8.52. Found: C, 60.76; H, 4.22; N, 8.46. IR (KBr, νmax/cm−1): 3058 (br, O–Hphenolic), 1624, 1587 (CNazomethine), 1374 (C–Ophenolic), 1254 (C–Oenolic), 1031 (N–N), 982 (VO). λmax (CH2Cl2)/nm (ε/dm3 mol−1 cm−1): 532 (5160), 334 (14100). 1H NMR (500 MHz, CDCl3, 298 K, δ/ppm): 7.67 (d, 1H, H-3, J = 8.5), 7.64 (d, 1H, H-5, J = 8.0), 6.82 (d, 1H, H-6, J = 8.0), 6.92 (d, 1H, H-11, J = 8.5), 7.50 (t, 2H, H-12, H-18, J = 8.0), 6.68 (t, 1H, H-13, J = 8.0), 7.32 (d, 1H, H-14, J = 8.0), 2.96 (s, 3H, 3 × H-15), 7.23–7.29 (m, 3H, H-17, H-19, H-22), 8.14 (d, 1H, H-21, J = 8.0), 7.91 (d, 1H, H-23, J = 4.5), 2.40 (s, 3H, 3 × H-25), 11.36 (s, 1H, O–Hphenolic). 51V NMR (500 MHz, 293 K, δ/ppm): −478.8 (CDCl3), −476.2 (DMSO-d6). ESI-MS (positive) in CH3CN: 494 (M + H+), 516 (M + Na+), 532 (M + K+).
2.3.3. [VVO(HL3)(hq)] (3). This compound was prepared following an identical method as described for compound 1 using the ligand H3L3 instead of H3L1. Yield: 0.42 g (82%). Anal. calc. for C25H20N3O6V: C, 58.95; H, 3.96; N, 8.25. Found: C, 58.78; H, 3.98; N, 8.21. IR (KBr, νmax/cm−1): 3445 (br, O–Hphenolic), 1624, 1589 (CNazomethine), 1369 (C–Ophenolic), 1253 (C–Oenolic), 1036 (N–N), 965 (VO). λmax (CH2Cl2)/nm (ε/dm3 mol−1 cm−1): 527 (4870), 337 (12450). 1H NMR (300 MHz, CDCl3, 298 K, δ/ppm): 7.48–7.51 (m, 2H, H-3, H-14), 7.23–7.31 (m, 4H, H-5, H-18, H-19, H-22), 6.87 (d, 1H, H-6, J = 8.7), 6.94 (d, 1H, H-11, J = 8.1), 7.67 (t, 1H, H-12, J = 8.1), 6.68 (t, 1H, H-13, J = 8.1), 2.95 (s, 3H, 3 × H-15), 8.15 (d, 1H, H-21, J = 8.1), 7.91 (d, 1H, H-23, J = 3.9), 3.87 (s, 3H, 3 × H-25), 11.34 (s, 1H, O–Hphenolic). 51V NMR (500 MHz, 293 K, δ/ppm): −476.1 (CDCl3), −474.1 (DMSO-d6). ESI-MS (positive) in CH3CN: 510 (M + H+), 532 (M + Na+), 548 (M + K+).
2.3.4. [VVO(HL4)(hq)] (4). This compound was prepared following an identical method as described for compound 1 using the ligand H3L4 instead of H3L1. Yield: 0.47 g (91%). Anal. calc. for C24H17ClN3O5V: C, 56.10; H, 3.33; N, 8.18. Found: C, 55.96; H, 3.48; N, 8.12. IR (KBr, νmax/cm−1): 3435 (br, O–Hphenolic), 1604, 1588 (CNazomethine), 1367 (C–Ophenolic), 1246 (C–Oenolic), 1036 (N–N), 981 (VO). λmax (CH2Cl2)/nm (ε/dm3 mol−1 cm−1): 556 (4710), 337 (13000). 1H NMR (300 MHz, CDCl3, 298 K, δ/ppm): 7.49–7.53 (m, 2H, H-3, H-14), 7.91 (dd, 1H, H-5, J = 8.7, 1.2), 6.87 (d, 1H, H-6, J = 8.7), 6.93 (dd, 1H, H-11, J = 8.1, 0.9), 7.68 (t, 1H, H-12, J = 8.1), 6.69 (dt, 1H, H-13, J = 8.1, 1.2), 2.94 (s, 3H, 3 × H-15), 7.22–7.32 (m, 3H, H-17, H-18, H-19), 8.16 (dd, 1H, H-21, J = 8.1, 1.2), 7.42 (dd, 1H, H-22, J = 8.1, 1.8), 7.79 (d, 1H, H-23, J = 2.4), 11.22 (s, 1H, O–Hphenolic). 51V NMR (500 MHz, 293 K, δ/ppm): −487.5 (CDCl3), −486.1 (DMSO-d6). ESI-MS (positive) in CH3CN: 514 (M + H+), 536 (M + Na+), 552 (M + K+).
2.4. Crystallographic data collection and refinement
X-ray quality single crystals of the H3L3 ligand were obtained from the slow evaporation of its acetone solution at room temperature while those of complexes 1–4 were obtained from the slow diffusion of n-hexane into the dichloromethane solution of the respective complex at room temperature. X-ray data for the H3L3 ligand and complexes 1, 2 and 4 were measured with MoKα radiation (λ = 0.71069 Å) at 150 K using the Oxford X-calibur CCD system while for complex 3 at 293 K using the Bruker APEX-II CCD system. Data analysis for 3 was carried out with Bruker SAINT program26 while for other compounds with the CRYSALIS program.27 Absorption corrections for 3 were carried out with SADABS program28 while for other compounds with the ABSPACK program.29 Structures were refined on F2 using the SHELXL97 program.30 Table S1† lists the key crystallographic parameters of H3L3 and complexes 1–4. CCDC 1406044–1406048 contains the supplementary crystallographic data for H3L3 and 1–4 complexes.†
2.5. Computational study
All calculations were performed with LANL2DZ31 effective core potentials and basis functions were used for the vanadium atom along with corresponding 6-311++G**32 and 6-31G33 basis set by using a B3LYP34 functional in the Gaussian 09 package.35 The geometry of the complexes was optimized starting with the geometries provided by the crystal structures of all four complexes 1–4. The Time Dependent Density Functional Theory (TD-DFT) method36 has been employed to calculate the possible excitation energies and intensities of all four complexes in the ground state optimized geometry. The 51V magnetic shielding was computed using B3LYP functional and 6-311++G** basis set. Calculations were carried out using either geometry optimized structures or the non-optimized X-ray geometry at the same level of theory as NMR parameter calculations, as specified in Table 4. The 51V chemical shifts are referenced to VOCl3. The VOCl3 was optimized using the same basis set. Electron affinity values of the complexes were calculated utilising crystal parameters by using B3LYP functional and 6-311++G** basis set. The DFT optimized geometries of ligand and complex molecules were used in docking. For docking studies the structural coordinates of CT-DNA were obtained from the protein data bank (PDB ID: 1BNA). Molecular docking for the four ligands and corresponding complexes were carried out using AutoDock 4.2.37 The conformation of the DNA was fixed and the complexes were allowed to change during molecular docking. In the course of docking −0.0001 gasteiger charge was for complex 1, 2, 3 and −0.0002 for 4. Thus the binding sites for the ligand molecules as well as their vanadium complexes in DNA were predicted. The data obtained from docking was examined by PyMOL (http://pymol.sourceforget.net/) software.38 Discovery Studio 4.1 Client and Chimera 1.10.1rc were used for visualization effects.
2.6. DNA binding studies
2.6.1. UV-vis spectral study. Absorption spectral titration experiments were performed by monitoring a 544 nm band for complex 1, 532 nm for complex 2, 527 nm band for complex 3 and 556 nm for complex 4 (which are the λmax values for the ligand-to-metal-charge-transfer transition band of the respective complexes). In each case the concentration of the complex was maintained at 10−5 M, while the concentration of the CT DNA was varied between the range of (0.1–1.0) × 10−5 M. An appropriate quantity of CT DNA was added to both the complex solution and the reference solution to eliminate the absorbance of DNA itself. From the absorption data, the intrinsic binding constant Kb was determined39 from a plot of [DNA]/(εa − εf) versus [DNA] using the eqn (1):| | |
[DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf)
| (1) |
where [DNA] is the concentration of DNA base pairs. The apparent extinction coefficients εa, εf and εb correspond to Aobsd/[complex] in the presence of DNA, in the absence of DNA and to fully bound DNA respectively. A plot of [DNA]/(εa − εf) versus [DNA] gave a slope of 1/(εb − εf) with intercept 1/[Kb(εb − εf)], so that Kb is obtained from the slope to intercept ratio.
2.6.2. Fluorometric study. In this binding experiment, 50 μL of a CT DNA (10−4 M) solution in Tris–HCl–NaCl buffer (5 × 10−2 mol Tris–HCl and 5 × 10−2 mol NaCl, pH = 7.2) was added to 2.0 mL of ethidium bromide (1.5 × 10−4 M) in the same buffer medium to get the maximum fluorescence intensity. Aliquots of 10−3 M stock solution of the complex in DMSO were added to the ethidium bromide bound CT DNA solution and the fluorescence was measured for each test solution after 2 hours of incubation at 37 °C. The solutions were excited at 515 nm (with excitation and emission slit 5 nm) and emission spectra were recorded from 520 to 700 nm. The solutions containing an equivalent amount of a CT DNA solution, complex 1–4 and buffer when excited at 515 nm did not exhibit any fluorescence emission. The fluorescence quenching experimental data were further analysed using the Stern–Volmer equation.40
2.6.3. 51V NMR study. In this binding experiment, initially, the 51V NMR spectra of a 0.5 mL of 20 mM DMSO-d6 solution of each of the complexes 1–4 were taken. Again the 51V NMR spectra of the respective solution was taken after the addition of 50 μL of 4 mM CT DNA solution in 10 mM Tris–HCl buffer (pH = 7.2).
2.6.4. Circular dichroism study. Circular dichroism (CD) spectra were obtained with a Jasco J-1500 spectropolarimeter with 200 μL sample containing CT DNA in 1 mM sodium phosphate buffer (pH = 7.2). CT DNA was incubated separately with all four vanadium complexes 1–4 for 3 h before obtaining the CD spectra. Data were expressed as degrees of ellipticity (h), in units of millidegrees (mdeg). The scan rate of 50 nm min−1 was maintained and 1 mm path length cuvette was used.
2.7. DNA cleavage study
With the help of agarose gel electrophoresis, influence of complexes on the cleavage of the supercoiled (SC) pUC19 was studied. pUC19 (500 ng) was incubated with 250 μM and 500 μM of each of the complexes 1–4 at 37 °C for 24 h in 20 μL reaction volume in 10 mM sodium phosphate buffer (pH = 7.2). Similar types of cleavage experiment was carried out using 250 μM of the four ligands in turn. The cleavage of pUC19 by the complexes was monitored on 1.25% (w/v) agarose gels run at 100 volts for 1.5 h in 1× TAE buffer and stained with ethidium bromide and the bands were visualised by UV light and agarose gel images were captured by the UVP GelDoc-It 310 gel documentation system. The formation of the nicked circular DNA and linearized DNA was checked by gel electrophoresis after incubation with 0.25–0.5 mM complexes of vanadium.
3. Results and discussion
3.1. Synthesis
Four hydrazone ligands (H3L1–4) obtained by the condensation of 2-hydroxybenzoylhydrazine with 2-hydroxyacetophenone and its 5-substituted derivatives have been utilised to synthesize the four mixed-ligand oxidovanadium(V) [VVO(HL1–4)(hq)] (1–4) complexes utilising 8-hydroxyquinoline (Hhq) as coligand in order to study the influence of substituents on the chemical property of vanadium as well as on the pharmacological activity of the complexes. The resulting complexes are shiny black in colour and soluble in CH2Cl2, CHCl3, CH3CN and highly polar solvents such as DMSO and DMF. These complexes are diamagnetic at 298 K indicating the +V state of vanadium and are non-electrolytic in nature.
3.2. Spectral characteristics
The spectral (IR, Mass, 1H NMR and UV-Vis) data of the ligands (except H3L1, as it has been recently reported in the literature25) and their corresponding mixed-ligand oxidovanadium(V) complexes 1–4 along with 51V NMR spectra are given in the Experimental section.
In the IR spectra of the ligands, two bands in the region 1627–1632 and 1613–1616 cm−1 are assigned to CO and CN stretching respectively. The phenolic O–H and N–H bands are observed respectively in the 3266–3452 and 2940–3083 cm−1 region. The N–N band is observed in the range 914–921 cm−1. Disappearance of characteristic CO and N–H bands of the ligands and the appearance of a new band in the 1247–1254 cm−1 region [assigned to ν(C–Oenolic)] in the vanadium complexes, indicate the formation of C–Oenolic moiety through enolisation of the –C(O)–NH– moiety followed by its coordination to vanadium through deprotonation.18b,41 The strong band near 1588 cm−1 is attributed to CN bond while the VO stretching band is observed in the 965–982 cm−1 region which is usual for similar type of vanadium complexes.1e,22 The N–N band of the complexes were observed in the 1027–1036 cm−1 region indicating a 109–122 cm−1 shift to a higher wave number upon complexation due to diminished repulsion between the lone pairs of adjacent nitrogen atoms.1e,22d,41 A sharp broad band observed in the 3058–3445 cm−1 region is likely to be associated with the intramolecularly H-bonded phenolic O–H group.
The electrospray ionisation (ESI) mass spectroscopy of the ligands (H3L2–4) and the complexes 1–4 have been recorded in acetonitrile solution. The ESI mass spectral data are summarised in the Experimental section. All the complexes show the molecular ion peak associated with a proton with 100% abundance.
The 1H NMR spectra of the free ligands (H3L2–4) were recorded in DMSO-d6 while that of complexes 1–4 were recorded in CDCl3 and data are given in the Experimental section. The spectra of all the ligands exhibit resonances in the regions δ 12.63–13.23 ppm and δ 11.54–11.60 ppm due to the phenolic O–H groups of the acetophenone and hydrazide part of the ligand respectively. The N–H resonance is observed at δ 11.90 ppm for H3L3 and at δ 11.88 ppm for H3L4 ligand. However, this resonance could not be observed for the H3L2 ligand. The disappearance of resonances in the range of δ 12.63–13.23 ppm in the complexes and the appearance of resonances in the region δ 11.22–11.36 ppm suggests that only the phenolic O–H group of the acetophenone moiety is involved in bonding with vanadium through deprotonation while the disappearance of the resonance peak ascribed to the amide (–CONH–) proton indicates that this moiety is also involved in bonding to V through the amide-O via enolisation followed by deprotonation (evidenced from the X-ray diffraction studies, vide infra).1e,22 All the aromatic protons of the complexes are observed in the region δ 6.68–8.16 ppm.
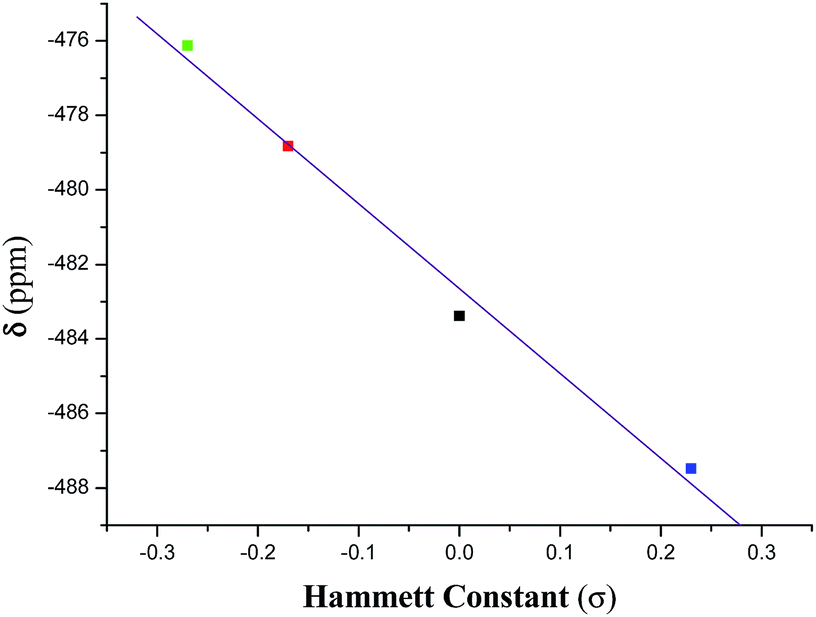
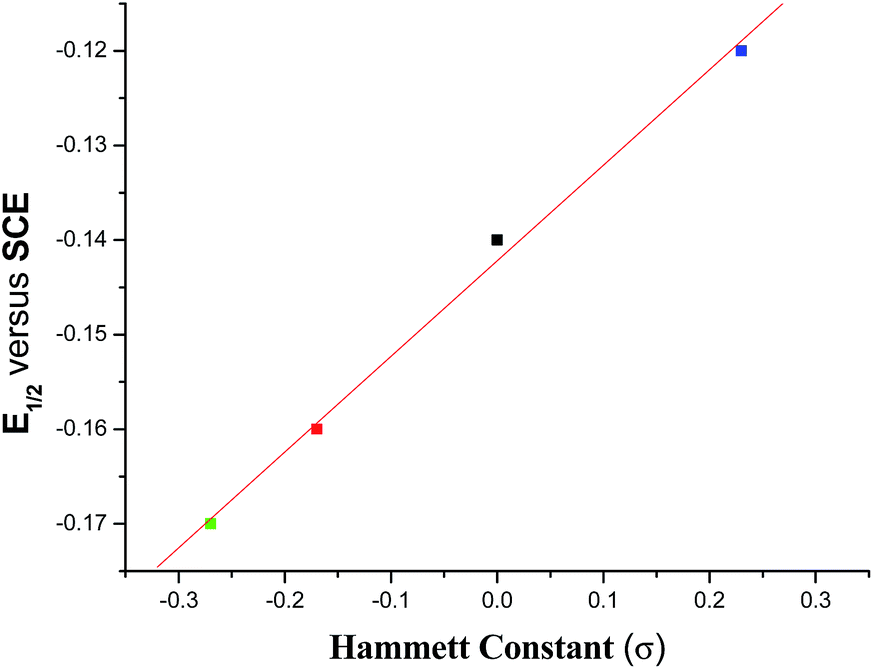
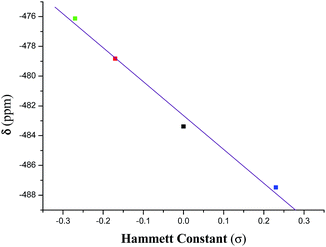
The 51V NMR spectrum of each of 1–4 in CDCl3 displays a singlet in the range −476.1 to −487.5 ppm (Fig. S1†) and in DMSO-d6 in the range −474.1 to −486.1 ppm (Fig. 11) which fall in the far upfield region of the standard VOCl3 as expected for oxygen and nitrogen rich environments42 and suggests their appreciable stability in solution. These chemical shifts are usual for oxidovanadium(V) complexes containing this type of mixed-ligand environment.22c,43 An analysis of the chemical shift (δ) data reveals that the introduction of an electron releasing CH3 group at the 5-position in the acetophenone ring (complex 2) resulted in a downfield chemical shift of ∼4.5 ppm with respect to the unsubstituted (complex 1). With the introduction of a more electron releasing OCH3 group (complex 3), a large downfield (by ∼7 ppm) chemical shift is observed while the introduction of the electron-withdrawing chloro substituent to the acetophenone ring (complex 4) resulted in an upfield shift (by ∼4 ppm). Such a trend is also supported by the DFT study (vide infra) of the 51V NMR spectra of the complexes. These chemical shift data reflect the varying electronic environment of the metal nucleus in these complexes containing different substituents. However, a linear relationship (eqn (2)) is observed on plotting the chemical shift (δ) values with the respective Hammett constants (σ) of the substituent [H (σ = 0.00), CH3 (σ = −0.17), OCH3 (σ = −0.27), Cl (σ = +0.23)] (Fig. 1):
| | |
δ (ppm) = −482.65 − 22.77 × σ
| (2) |
 |
| | Fig. 1 Plot of δ versus Hammett constant (σ) for complexes 1–4. | |
This correlation (with an r value of 1.0) indicates that the dependence of δ value on the substituent (i.e., dδ/dσ) is −22.77.
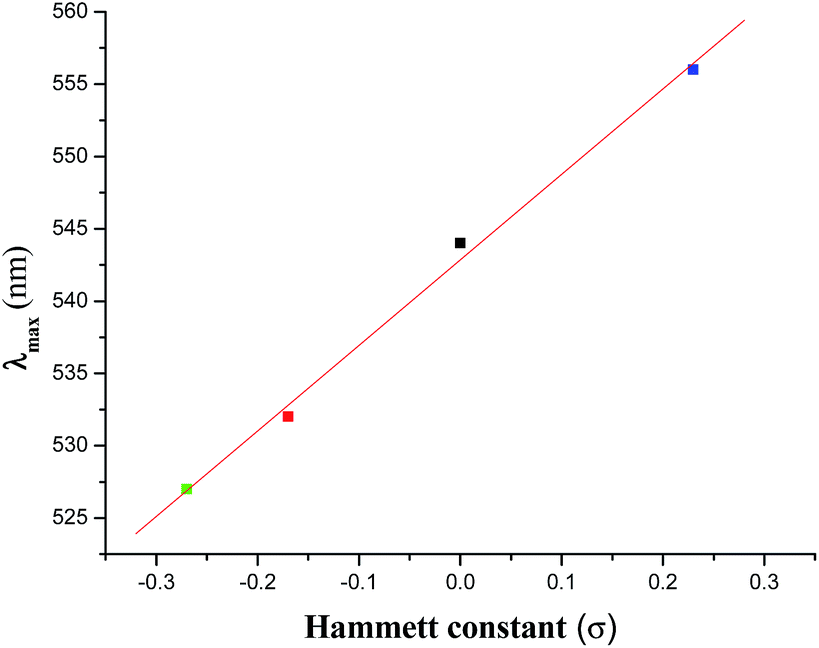
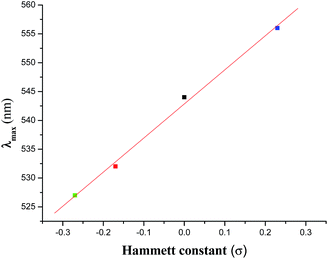
Electronic spectra of the complexes were recorded in CH2Cl2 solution. Complexes exhibit two intense transitions in the 300–800 nm regions (Table 1), one in the 527–556 nm regions and the other in the range 332–337 nm (Fig. S2†). These are most likely due to ligand-to-metal-charge transfer (LMCT) transitions (more specifically from the Ph–O of the hq− to V+5, supported by DFT calculations, vide infra) and the intra-ligand π → π* transition respectively. The LMCT transitions are usual for similar type of mixed-ligand oxidovanadium(V) complexes.1e,22 Like the chemical shift (δ) value in the 51V NMR spectra, the λmax value of the LMCT transition also exhibits a linear relationship (eqn (3)) with the σ value of the substituent (Fig. 2).
| | |
λmax (nm) = 542.86 + 59.14 × σ
| (3) |
Table 1 Electronic spectral and electrochemicala data at 298 K of the complexes 1–4 in CH2Cl2 solution
| Complex |
λmax/nm (ε/dm3 mol−1 cm−1) |
(E½)b/V (ΔEp/mV)c |
| At a Pt electrode; supporting electrolyte: Bu4nN+ClO4− (TBAP ∼ 0.1 M); scan rate: 50 mV s−1 reference electrode: SCE; solute concentration: ∼10−3. E½ is calculated as the average of anodic (Eap) and cathodic (Ecp) peak potentials. ΔEp = Eap − Ecp. |
| [VVO(HL1)(hq)] (1) |
544(4860), 332(15000) |
−0.14(170) |
| [VVO(HL2)(hq)] (2) |
532(5160), 334(14100) |
−0.16(180) |
| [VVO(HL3)(hq)] (3) |
527(4870), 337(12450) |
−0.17(180) |
| [VVO(HL4)(hq)] (4) |
556(4710), 337(13000) |
−0.12(180) |
 |
| | Fig. 2 Plot of λmax versus Hammett constant (σ) for complexes 1–4. | |
This correlation (with an r value of 1.0) indicates that the dependence of the λmax value on the substituent (i.e., dλmax/dσ) is 59.14.
3.3. Crystal structures
The molecular structure of H3L3 ligand and all the four complexes reported here have been determined by X-ray diffraction analysis and the crystal and refinement data are presented in Table S1.†
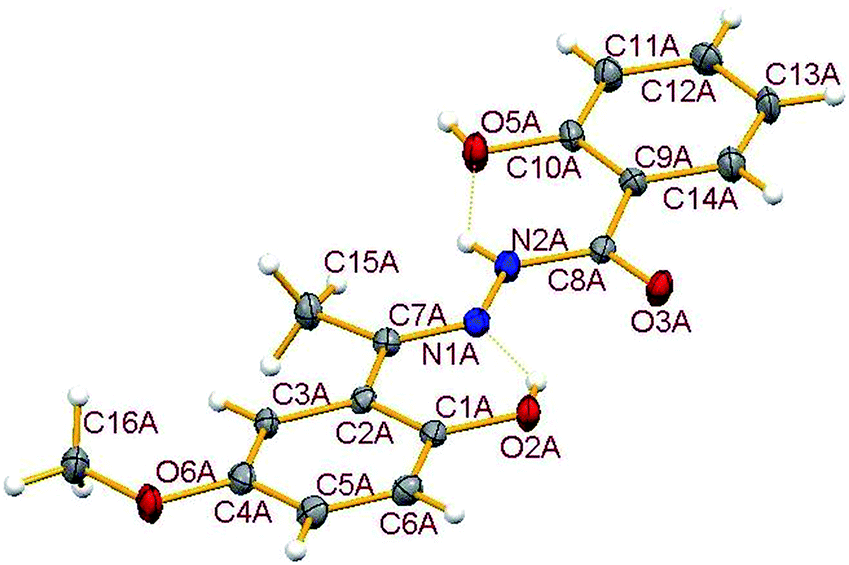
3.3.1. Description of the structure of H3L3. The crystal structure of the hydrazone ligand H3L3, a condensed product of 2-hydroxybenzoylhydrazide and 2-hydroxy-5-methoxy-acetophenone contains two molecules in the asymmetric unit (Fig. S3†), called here A and B, which have similar geometries and closely matching dimensions, together with one solvent acetone molecule. For clarity only molecule A is shown in Fig. 3. Important dimensions are compared in Table S2.† The formation of the hydrazone ligand is confirmed from the double bond nature of C(7)–N(1) bond in both A and B. The amide form of the free hydrazone ligand is evident from the single bond nature of the C(8)–N(2) bond and also from the double bond nature of C(8)–O(3) bond, though it undergoes enolisation on complexation. Two intramolecular hydrogen bonds, involving N(2)–H with O(5) and O(2)–H with N(1) are observed in both molecules. In addition, the two molecules are connected via two hydrogen bonds: O(5A)–H(5A)⋯O(3B) (1 + x, −1 + y, z) and O(5B)–H(5B)⋯O(3A) (−1 + x, y, z). Dimensions for hydrogen bonds are given in Table S3.†
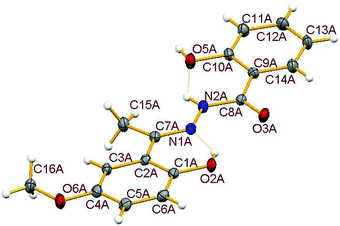 |
| | Fig. 3 Molecular structure of H3L3-A showing the atom numbering scheme with ellipsoids at 30% probability. Hydrogen bonds are shown as dotted lines. Molecule B which has equivalent dimensions and the solvent acetone are omitted for clarity. Selected geometric parameters (Å): C(7A)–N(1A) 1.292(4), N(1A)–N(2A) 1.370(3), C(8A)–N(2A) 1.351(3), C(8A)–O(3A) 1.243(4). | |
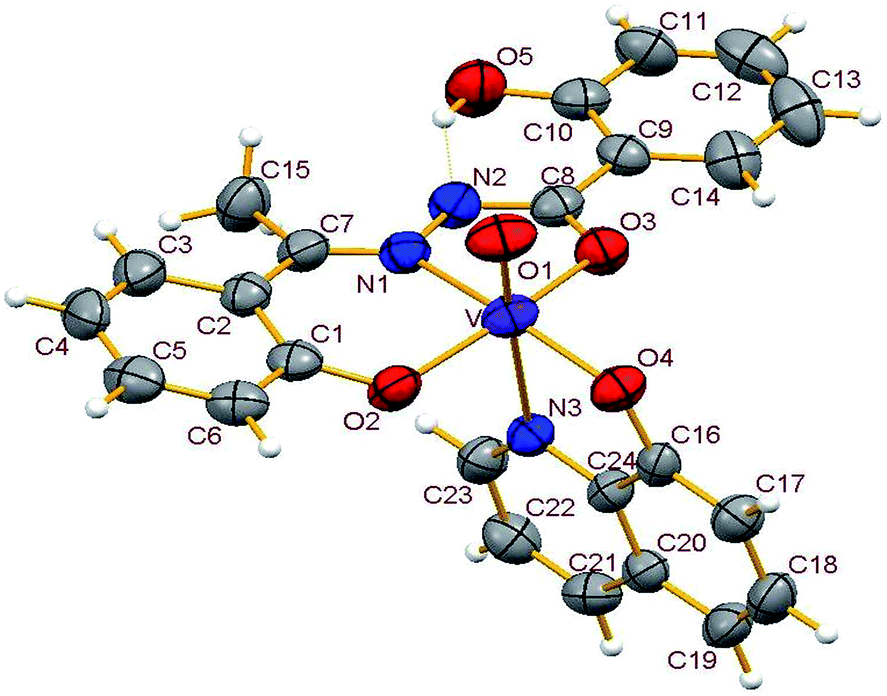
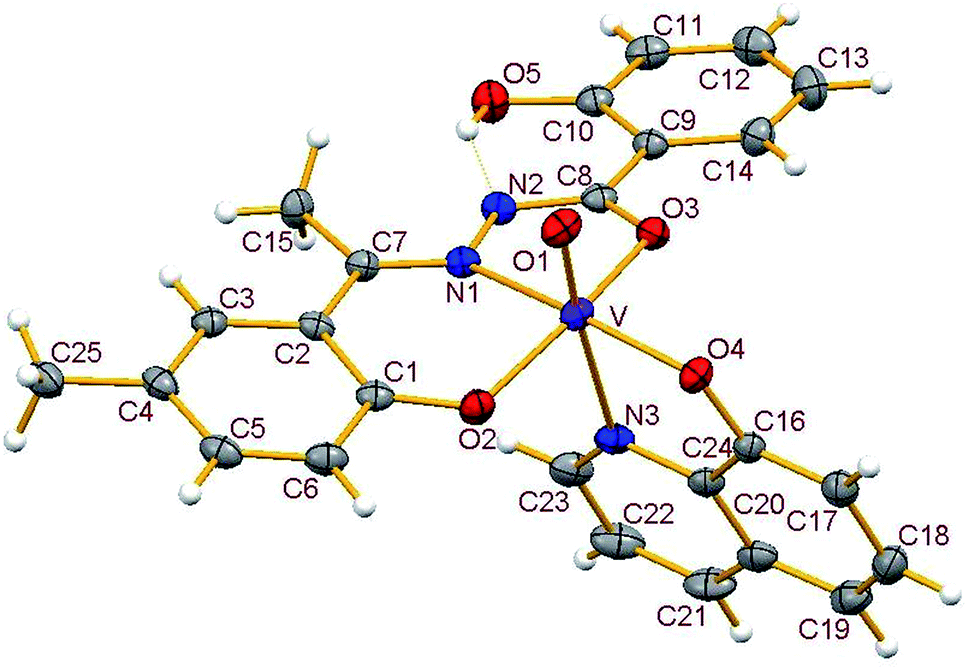
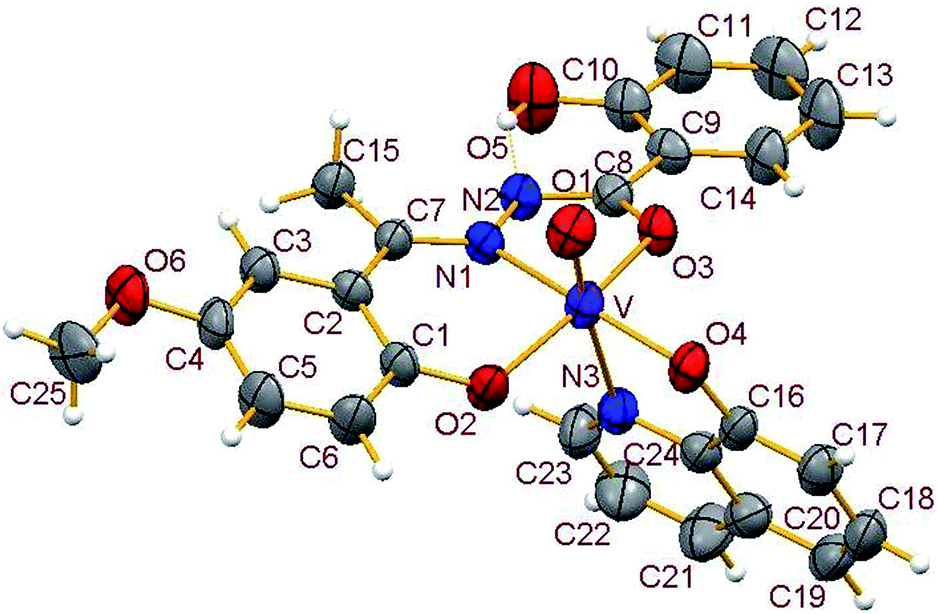
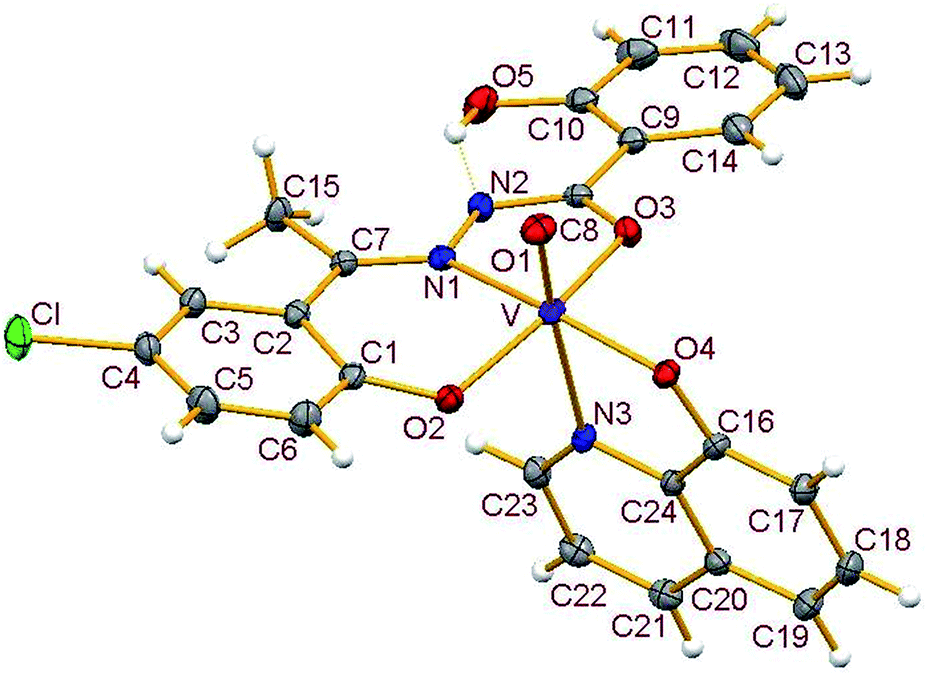
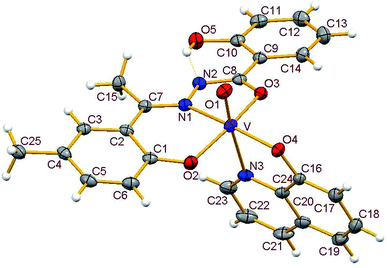
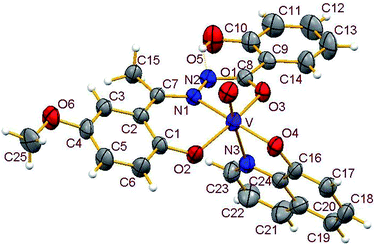
3.3.2. Description of the structure of 1–4. Crystals of complexes 1 and 3 belong to the monoclinic crystal system, in space group P21/c while complexes 2 and 4 belong to the triclinic crystal system, in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Complexes 1–4 have similar structures. Identical atom-labelling schemes have been adopted for all the four structures for easy comparison of their relevant metrical parameters. Molecular views of 1–4 are displayed in Fig. 4–7, respectively.
. Complexes 1–4 have similar structures. Identical atom-labelling schemes have been adopted for all the four structures for easy comparison of their relevant metrical parameters. Molecular views of 1–4 are displayed in Fig. 4–7, respectively.
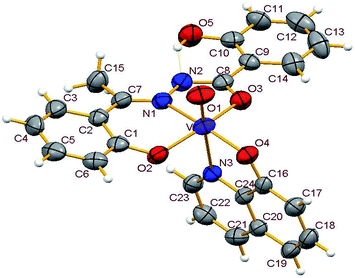 |
| | Fig. 4 ORTEP view of complex 1 with ellipsoids at 30% probability. Hydrogen bonds are shown as dotted bonds. | |
 |
| | Fig. 5 ORTEP view of complex 2 with ellipsoids at 30% probability. Hydrogen bonds are shown as dotted bonds. | |
 |
| | Fig. 6 ORTEP view of complex 3 with ellipsoids at 30% probability. Hydrogen bonds are shown as dotted bonds. | |
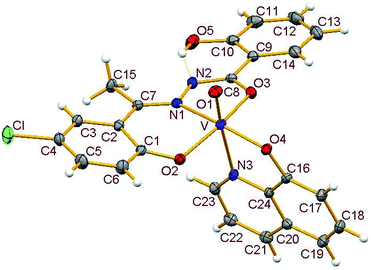 |
| | Fig. 7 ORTEP view of complex 4 with ellipsoids at 30% probability. Hydrogen bonds are shown as dotted bonds. | |
All four complexes consist of a neutral unit composed of a central vanadium ion in a distorted octahedral environment surrounded by one tridentate dinegative hydrazone ligand bound in a meridional manner (mer-ONO donor set in the enol form), one bidentate mononegative 8-hydroxyquinoline (ON donor set) occupying an equatorial and an axial position along with an oxido ion at the other axial position indicating the +V oxidation state of vanadium (also confirmed by magnetic susceptibility measurements). Selected bond lengths and bond angles in the four metal coordination spheres are compared in Table 2. There are also two other potential donor sites in the coordinated hydrazone ligand viz, phenolic-O(5) and the other imine-N(2) which are not coordinated due to their non-planarity with the three donor sites. However, in all structures the hydrogen atom on O5 is directed towards N2 forming an intramolecular hydrogen bond. Dimensions in 1 are typical with O(5)⋯N(2) 2.599(4) Å, H(5)–N(2) 1.88 Å and O(5)–H(5)⋯N(2) 145.6° and those for other complexes are given in Table S4.†
Table 2 Selected bond lengths (Å) and angles (°) in complexes 1–4
| |
1 |
2 |
3 |
4 |
| V–O1 |
1.593(3) |
1.592(2) |
1.594(3) |
1.584(2) |
| V–O2 |
1.836(3) |
1.842(2) |
1.857(3) |
1.855(2) |
| V–O3 |
1.952(3) |
1.978(2) |
1.968(3) |
1.951(2) |
| V–O4 |
1.833(3) |
1.850(2) |
1.851(3) |
1.843(2) |
| V–N1 |
2.111(3) |
2.087(2) |
2.117(4) |
2.100(2) |
| V–N3 |
2.378(3) |
2.336(2) |
2.367(4) |
2.339(2) |
| O1–V–O2 |
100.49(14) |
100.57(8) |
100.34(16) |
98.55(9) |
| O1–V–O3 |
99.78(14) |
99.95(8) |
100.67(15) |
99.44(9) |
| O1–V–O4 |
101.39(14) |
99.67(8) |
100.27(15) |
98.55(9) |
| O2–V–O3 |
152.21(12) |
151.87(7) |
149.97(15) |
153.88(8) |
| O2–V–O4 |
102.10(12) |
102.62(7) |
103.72(13) |
103.16(8) |
| O3–V–O4 |
92.27(12) |
92.66(7) |
92.15(12) |
92.73(8) |
| O1–V–N1 |
96.88(14) |
96.79(8) |
95.14(15) |
96.68(9) |
| O2–V–N1 |
82.74(12) |
82.82(17) |
82.76(13) |
82.98(8) |
| O3–V–N1 |
76.13(12) |
75.80(7) |
75.13(12) |
75.71(8) |
| O4–V–N1 |
159.82(12) |
161.34(7) |
161.64(13) |
160.61(8) |
| O1–V–N3 |
177.15(13) |
175.34(8) |
175.04(15) |
175.75(9) |
| O2–V–N3 |
80.84(11) |
82.84(7) |
82.86(14) |
82.59(8) |
| O3–V–N3 |
79.81(11) |
77.93(7) |
76.38(13) |
81.23(7) |
| O4–V–N3 |
75.83(11) |
76.38(7) |
76.01(14) |
76.21(7) |
| N1–V–N3 |
86.78(11) |
86.75(7) |
87.98(14) |
86.54(8) |
Thus, the vanadium atom is bonded to four oxygen atoms and two nitrogen atoms to form a distorted octahedral geometry. One phenolate O(2), one amide O(3) (in the enol form) and the imine nitrogen N(1) of the doubly deprotonated meridionally disposed (HL)2− ligands along with the phenolic oxygen O(4) of the deprotonated 8-hydroxyquinoline (hq−) constitute the equatorial plane with r.m.s. deviations of 0.015, 0.032, 0.055, 0.001 Å from which the vanadium atom is displaced by 0.326(2), 0.312(1), 0.321(1), 0.293(1) Å towards the axial terminal O(1) atom in the four structures respectively. The second axial position is occupied by the pyridine nitrogen N(3) of hq−. These hydrazone ligands generate a six-membered and a five-membered chelate ring at the vanadium center with the corresponding bite angles being in the ranges 82.74(12)–82.98(8)° and 75.13(12)–76.13(12)° respectively. The atoms in the five-membered ring are closely coplanar with a r.m.s. deviations of 0.018, 0.027, 0.054, 0.035 Å respectively while the six-membered ring is folded. The C–O, C–N and N–N bond lengths in the five-membered chelate ring are consistent with the enolate form of the amide. The atoms in the third chelate ring, [V–N(3)–C(24)–C(16)–O(4)], formed by the hq− are also approximately coplanar (with r.m.s. deviations of 0.006, 0.029, 0.042, 0.029 Å) with bite angles in the range 75.83(11)–76.38(7)° and this plane is almost perpendicular to the equatorial plane intersecting at angles of 89.5(1), 86.1(1), 85.0(1), 88.2(1)° respectively.
The shortest and longest bonds formed by the metal atom are respectively vanadium to terminal oxido group [i.e., VO(1)] and V–N(3), which lying trans to VO(1) is lengthened due to the trans influence of the oxido atom. The VO(1) (oxido), V–O(2)/V–O(4) (phenolato), V–O(3) (amido/enolato) bond lengths are well within the range reported for VO3+ complexes with carbonic acid hydrazide ligands and Hhq coligand.1e,22 The two phenolate bonds have similar lengths. These four vanadium–oxygen bond lengths in complexes are: oxo < phenolate < amide probably due to the order of O → V π-bonding.
3.4. Electrochemical results
The cyclic voltammograms of 1–4 each display one quasi-reversible wave (Fig. S4†) in CH2Cl2 with the E½ value near −0.15 V (Table 1) which are usual for similar type of complexes.1e,22 Practically no change in peak positions was observed but there was a slight change in peak heights on changing the scan rate (Fig. S5†). A scrutiny of the E½ values reveals that it decreases with increasing electron releasing property of the substituent in the acetophenone ring while it is increased by an electron withdrawing group. This trend is expected from a consideration of the electron density at the V-centre. A plot of E½ (V) value versus σ of the respective substituent reveals the following (eqn (4)) linear relationship with r value of 1.0 (Fig. 8).| | |
E½ = −0.14 + 0.10 × σ
| (4) |
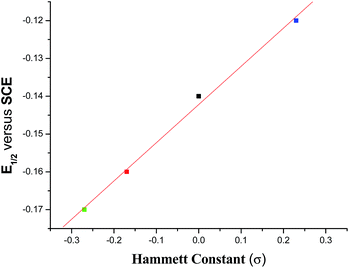 |
| | Fig. 8 Plot of E½ (V) versus Hammett constant (σ) for complexes 1–4. | |
By eliminating σ from eqn (3) and (4), one can obtain the following relationship (eqn (5)) correlating λmax (nm) with E½ (V)
| | |
λmax = 625.65 + 591.40 × E½
| (5) |
In fact, an almost similar relationship (eqn (6)) with an r value of 0.99 is obtained on plotting λmax (nm) versus the respective E½ (V) value of the complexes (Fig. S6†).
| | |
λmax = 626.00 + 584.74 × E½
| (6) |
3.5. The stability of vanadium complexes
The stability of molecules can be determined from the frontier molecular orbitals i.e., the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). HOMO and LUMO energies of four complexes calculated via DFT using two different basis sets for the ligands namely 6-311++G** and 6-31G are listed in Table 3. The HOMO–LUMO separation energies can be used to describe the kinetic stability and reactivity pattern of the complexes. A lower value of separation energy indicates higher reactivity and lower kinetic stability of the molecule. According to Pearson,44 the LUMO–HOMO separation energy represents the chemical hardness (η, corresponds to ΔE/2, where ΔE is the energy difference between the LUMO and HOMO) which is a reliable reactivity parameter able to predict the stability of a molecule. The maximum hardness principle45 states that the most stable molecule has the maximum η value (Table 3). It is observed from the calculations that the unsubstituted hydrazone complex 1 has a higher value of HOMO–LUMO energy gap; hence it is more stable than substituted hydrazone complexes. The complex 3 containing the most electron releasing group on the acetophenone moiety of the hydrazone ligand has the highest reactivity as indicated by the lowest HOMO–LUMO energy gap. It is noted that the energy gap in complex 4 (containing an electron withdrawing Cl atom) is comparable to complex 2 (containing an electron releasing CH3 group), however, the reason for this is not clear. It is also observed from Table 3 that the energy of the LUMO for the complexes follows the order: 4 < 1 < 2 < 3 which explains the reverse order of E½ values. The electron affinity values of the complexes (Table 3) are positive and are compatible with the negative E½ values. An analysis of electron affinity values indicate that the complex 3 has the least tendency to be reduced followed by the complexes 2, 1, and 4 respectively. Such a trend is also observed in their respective E½ values (vide supra).
Table 3 Determination of stabilitya of complexes 1–4 at the B3LYP level using different basis sets
| Complex |
6-311++G** |
EA (eV) |
6-31G |
| EH |
EL |
ΔE |
η |
EH |
EL |
ΔE |
η |
| EH and EL represent the energy (in eV) of HOMO and LUMO respectively. EA represents the electron affinity value in eV. |
| 1 |
−6.078 |
−3.335 |
2.743 |
1.371 |
7.83 |
−5.854 |
−3.003 |
2.851 |
1.425 |
| 2 |
−5.998 |
−3.289 |
2.709 |
1.354 |
8.29 |
−5.796 |
−2.964 |
2.832 |
1.416 |
| 3 |
−5.855 |
−3.258 |
2.597 |
1.298 |
8.34 |
−5.688 |
−2.946 |
2.742 |
1.371 |
| 4 |
−6.169 |
−3.465 |
2.704 |
1.352 |
7.01 |
−5.995 |
−3.181 |
2.814 |
1.407 |
The B3LYP/LANL2DZ/6-311++G** and 6-31G optimized geometrical parameters of 1–4 are in reasonable agreement with the values obtained from the X-ray crystallographic data. Some selected optimized bond lengths are shown in Table S5.† The V–O(1) (oxido) distances calculated by the 6-31G basis set are comparatively much closer to the experimental value than the values obtained by 6-311++G** basis set. On the other hand, other V–O distances i.e., V–Ophenolate [V–O(2) and V–O(4)] and V–Oamide [V–O(3)] are predicted more accurately by the later set. However, in both cases, the relatively long bond viz., V–N(1) is overestimated by 0.01–0.04 Å while the more long V–N(3) bond is calculated to be too long by ∼0.20 Å. The V–O(2) and V–O(4) bond lengths, determined from DFT optimized data indicates that these bond lengths are strongly dependent on the electronic effect of para substituents in the hydrazone ligands. The lengthening of V–O(2) bond is observed when hydrazone contains an electron withdrawing group and the reverse is true with an electron releasing group, while the V–O(4) bond length values follow the expected reverse order.
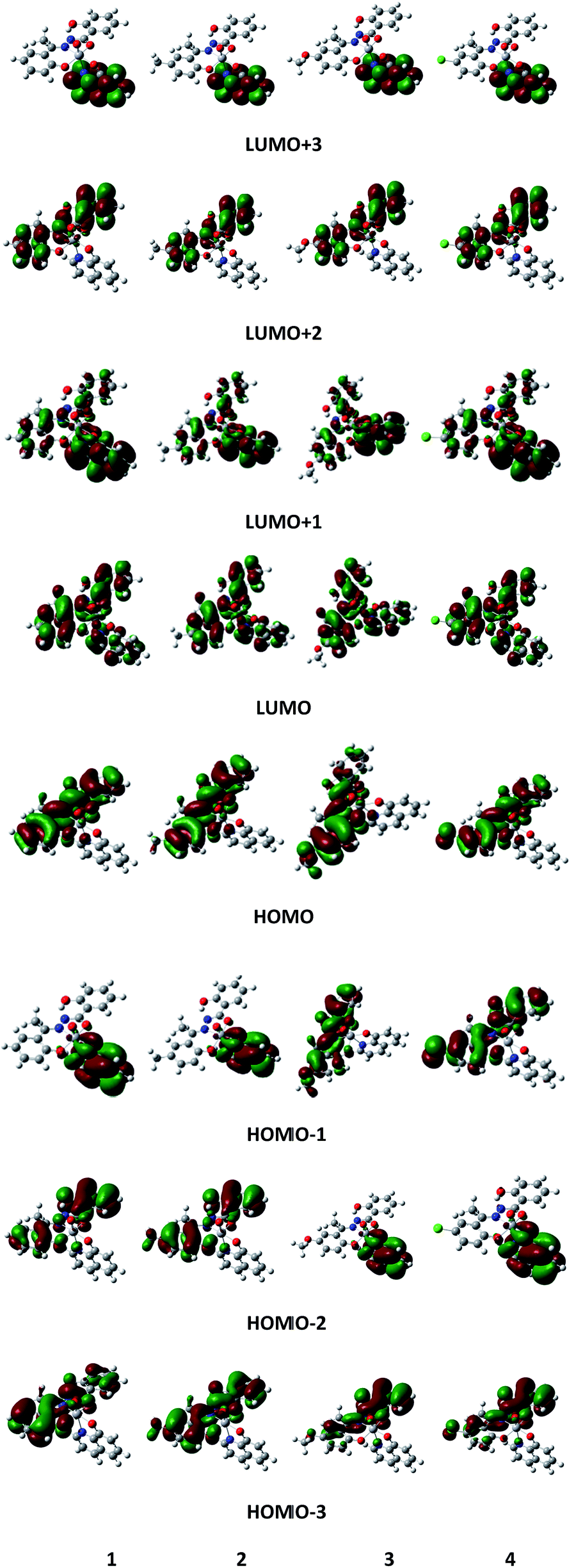
The MO pictures of several frontier orbitals of all complexes in their ground state optimized geometries are shown in Fig. 9. The vertical excitation energies and oscillator strengths of lowest few excited singlets of all complexes obtained from TD-DFT calculations by using 6-311++G** basis function at the corresponding ground state geometries are given in Table S6.† For 1, the largest oscillator strength has been noted for S3 at 487 nm compared to observed absorption maximum at 544 nm. The S3 state is dominated by the HOMO−1 → LUMO excitation along with the HOMO−3 → LUMO transition. The excitation energy of the S2 state corresponds to 508 nm with oscillator strength f = 0.0028 and it is mainly characterized by the HOMO−2 → LUMO transition. For 2, the maximum value of f (f = 0.2027) is obtained at 486 nm (S3) (cf. the experimentally observed peak at 532 nm). The excitation energy of second excited singlet (S2) is 511 nm. Both S2 and S3 are characterized as a HOMO−1 → LUMO transition. For 3, the maximum value of oscillator strength (f = 0.1898) is obtained at 486 nm (S3) compared to observed absorption maximum at 527 nm. The second excited singlet (S2) is 528 nm. S2 is characterized by HOMO−1 → LUMO while S3 is characterized by the HOMO−2 → LUMO transition. For complex 4, the maximum oscillator strength (f = 0.2111) is noted for S3 at 489 nm compared to the experimentally observed 556 nm and is characterized as a HOMO−2 → LUMO transition. The second excited singlet S2 is 519 nm which is due to a HOMO−1 → LUMO transition. For all complexes, the computed absorption maximum is at higher energy end than the corresponding experimental value. However, from the frontier molecular orbital diagrams of the complexes (Fig. 9), it is evident that an LMCT transition is occurring from hq− ligand to V as the HOMO is localised mainly on the hydroxyquinoline ring while the LUMO is localised on a 3d orbital of vanadium atom.
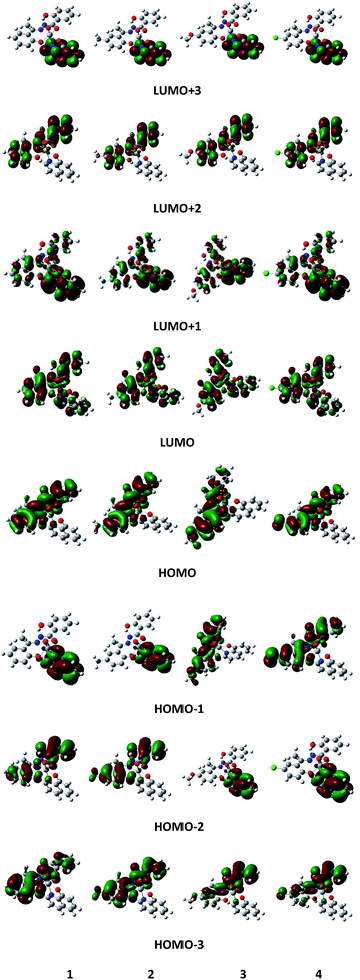 |
| | Fig. 9 Schematic diagram of selected frontier orbitals for complexes 1–4 in their ground state geometries. | |
DFT calculation reveals that the small HOMO–LUMO energy gap would produce downfield chemical shifts.42 When the hydrazone ligand is associated with an electron donating group, the HOMO–LUMO energy gap decreases (Table 3) and the 51V NMR spectra of the resulting complexes shows a downfield shift while with an electron withdrawing group the reverse is true (Table 4). The theoretical values particularly using DFT optimized parameters nicely match the experimental values.
Table 4 Experimental (in CDCl3 solution) and computed (by 6-311++G** basis functions) 51V parameters of 1–4 complexes
| Complex |
δxx = δref − σxx |
δyy = δref − σyy |
δzz = δref − σzz |
δiso = (δxx + δyy + δzz)/3 |
δ (experimental) |
| (Using DFT optimized parameters) |
| 1 |
−275.0 |
−363.4 |
−808.2 |
−482.2 |
−483.4 |
| 2 |
−265.3 |
−343.7 |
−815.7 |
−474.9 |
−478.8 |
| 3 |
−272.8 |
−324.6 |
−819.2 |
−472.2 |
−476.5 |
| 4 |
−265.4 |
−342.7 |
−794.6 |
−467.6 |
−487.5 |
|
| (Using X-ray crystallographic parameters) |
| 1 |
−222.0 |
−341.8 |
−848.9 |
−470.9 |
−483.4 |
| 2 |
−220.7 |
−314.8 |
−862.6 |
−466.0 |
−478.8 |
| 3 |
−227.8 |
−299.7 |
−868.6 |
−465.4 |
−476.5 |
| 4 |
−221.6 |
−309.7 |
−845.7 |
−459.0 |
−487.5 |
3.6. DNA binding studies
The DNA binding ability of the complexes 1–4 has been studied by various spectroscopic techniques which are described below.
3.6.1. Electronic absorption spectral studies. Electronic absorption spectroscopy is one of the most useful techniques to examine the binding of the metal–ligand complexes with DNA. The electronic spectra of all the complexes 1–4 in the presence and absence of DNA were studied. The significant modification of the UV absorption band of all complexes on addition of increasing amounts of DNA indicates the binding with DNA. With the addition of an increasing amount of CT DNA to each of the 1–4 complexes, a decrease in molar extinction coefficient of the LMCT transition band46 with hypochromism associated with a significant red shift (7–21 nm) is observed for complexes 1–3 whereas hypochromism with a blue shift of 5 nm is observed for complex 4 (Fig. S7†), suggesting a strong binding ability of complexes 1–3 to DNA. Hypochromism results from the contraction of DNA helix as well as conformational changes to DNA47,48 while hyperchromism results from a change in the secondary structure of a DNA double helix. This observation has been further supported by quantitative studies. In order to quantitatively compare the binding strength of all complexes with CT DNA, the intrinsic binding constant (Kb) values were calculated following eqn (1) and were found to be (3.89 ± 0.2) × 105, (9.8 ± 0.2) × 105, (1.7 ± 0.2) × 106 and (1.9 ± 0.2) × 105 M−1 for 1–4 respectively. The Kb values follow the order: 3 > 2 > 1 > 4, suggesting that the presence of an electron donating group in the aromatic ring increases the complexes' binding ability with DNA base pairs. The amount of hypochromism along with red shift and DNA binding constants values indicate that complexes 1 and 4 bind the DNA at the minor groove while complexes 2 and 3 bind the DNA through partial intercalation.25,46–50The change in Gibbs free energy observed (ΔGobs) for DNA binding, can be calculated from standard Gibbs equation (ΔGobs = −RTlnKb). The ΔGobs values were derived to be −7.93, −8.49, −8.81 and −7.50 kcal mol−1 at 37 °C for complexes 1–4 respectively. These experimentally calculated ΔGobs values are found to be close to the values of ΔGtheo obtained from molecular docking (vide infra).
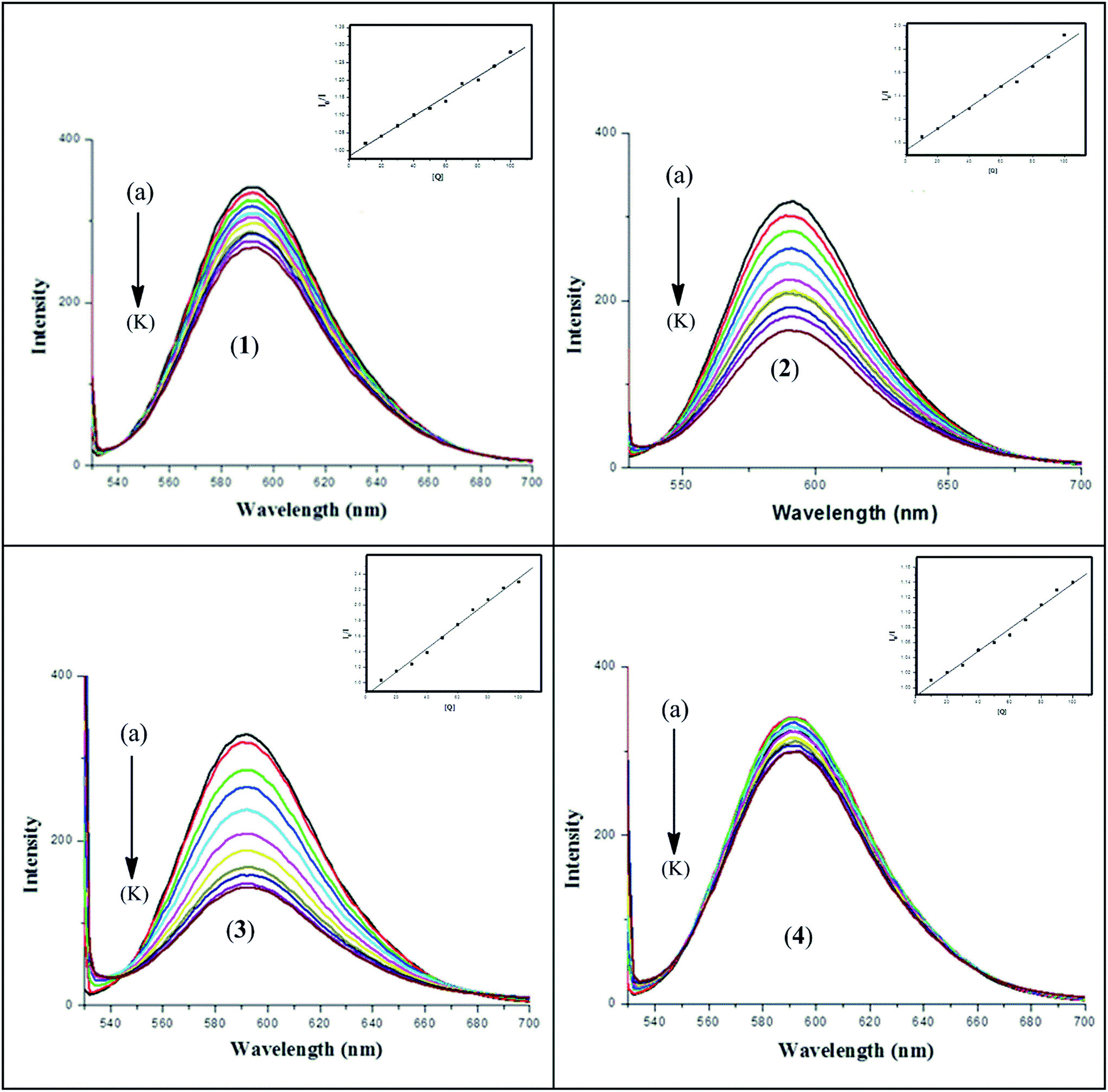
3.6.2. Fluorescence emission study for DNA interaction. The results obtained from spectrofluorometric titrations indicate that all four complexes 1–4 effectively bind to DNA. In order to confirm the mode of binding and to compare the binding ability of the metal complexes with DNA, we have carried out an ethidium bromide (EB) displacement experiment. In competitive ethidium bromide (EB) binding studies, complexes 1–4 was added to DNA pre-treated with EB followed by measurement of DNA-induced emission intensities of EB (Fig. 10). To quantify the affinity of the complexes to DNA, the Stern–Volmer equation40 (I0/I = 1 + Kav[Q]) was employed. The binding constant Kav was obtained from the slope of the plot of I0/I vs. [Q]. The Kav values were found to be (2.89 ± 0.2) × 105, (9.75 ± 0.2) × 105, (1.80 ± 0.2) × 106 and (1.52 ± 0.1) × 105 M−1 at 37 °C for complexes 1–4 respectively. An appreciable decrease in the emission intensities was observed upon the addition of the complexes to the EB–CT-DNA system, which was due either to the intercalation or groove binding of the complexes to DNA base pairs that replace some EB molecules from the EB–CT-DNA system.51 ΔGav values were derived to be −7.74, −8.49, −8.87 and −7.35 kcal mol−1 for complexes 1–4 respectively. This observation also supports the results obtained from the electronic absorption spectrophotometric analysis and molecular docking (vide infra) that complexes 1 and 4 bind with DNA by groove binding and complexes 2 and 3 by partial intercalation. The Kav values for the ligand–DNA system are found to be (7.34 ± 0.2) × 104, (1.1 ± 0.2) × 105, (1.05 ± 0.2) × 105 and (8.40 ± 0.2) × 104 M−1 for H3L1–4 respectively which are reasonably lower than the respective metal complexes (Fig. S8).
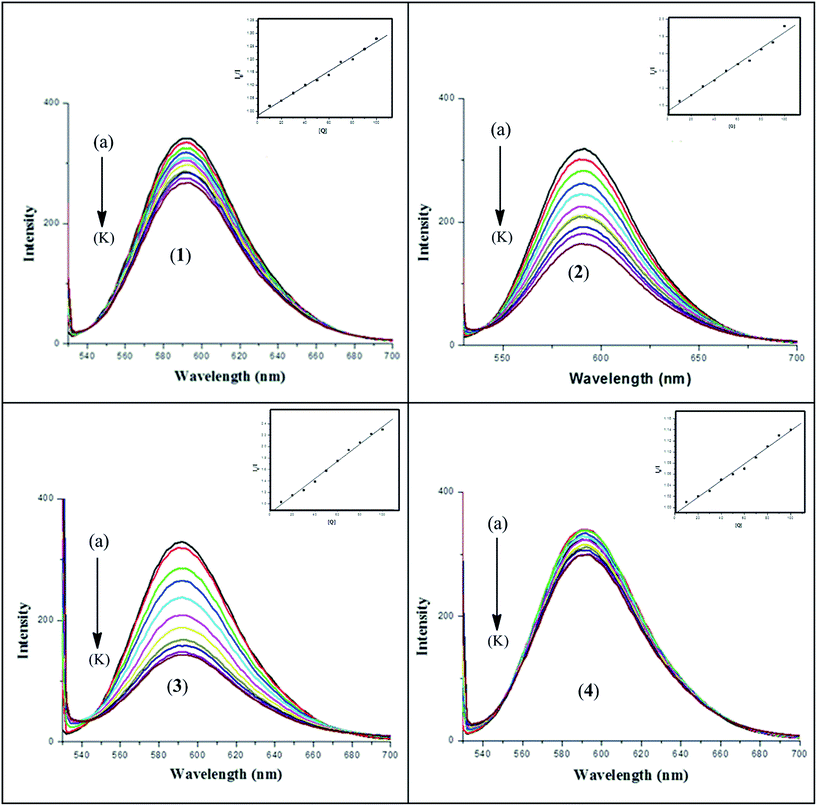 |
| | Fig. 10 Fluorescence spectra of (a) EB + 10−4 M DNA control and (b)–(k) EB + DNA + (1–10) × 10−5 M each of complex 1–4. The arrow shows that the intensity decreases with the increasing concentration of complexes 1–4. [Inset: Stern–Volmer plot for the quenching of fluorescence of the ethidium bromide (EB)–DNA complex caused by complexes 1–4.] | |
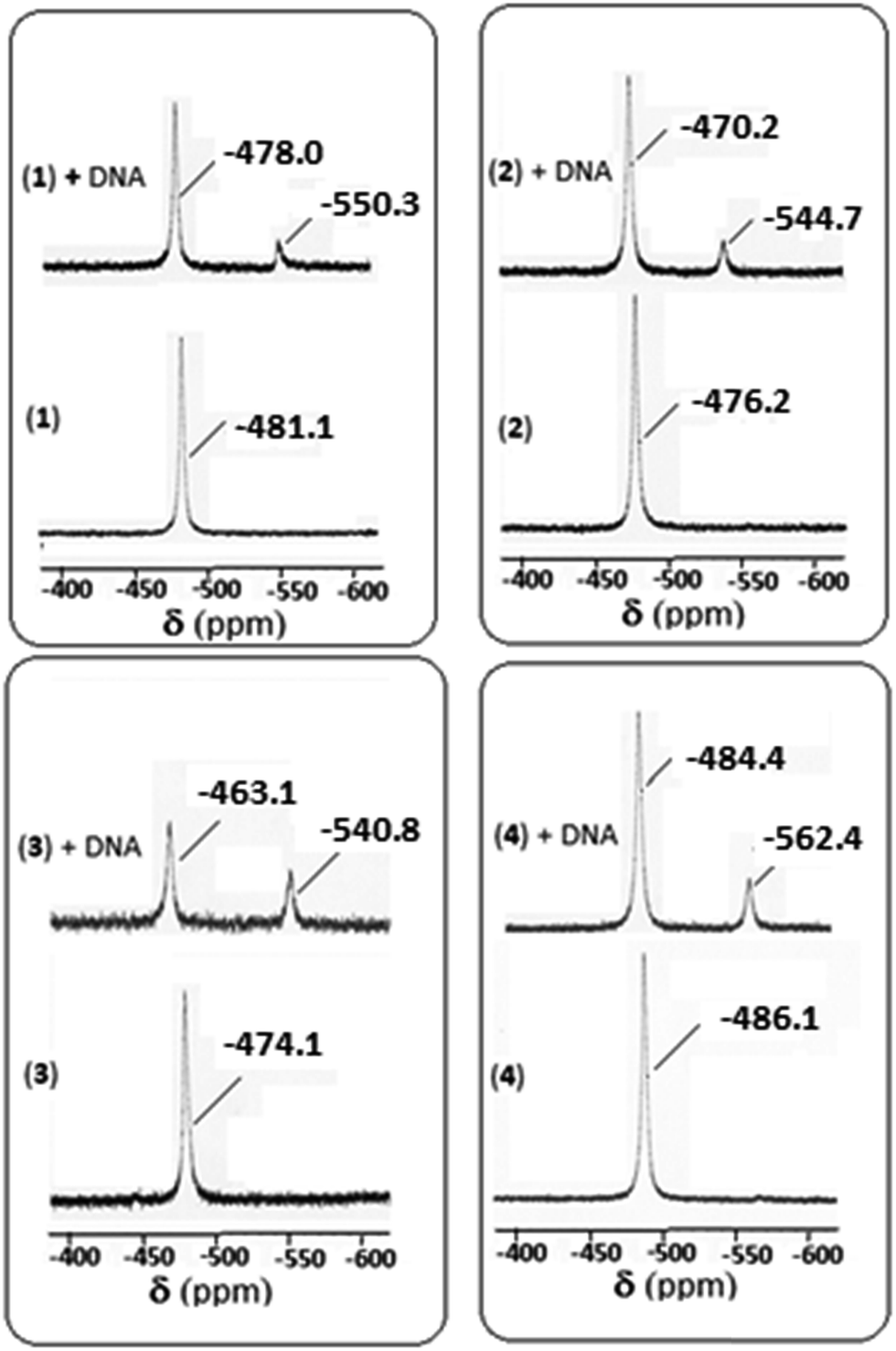
3.6.3. 51V NMR spectral study for DNA interaction. 51V NMR spectral study is also an important tool for studying the interaction with DNA. In the absence of DNA, the 51V NMR spectrum of each of 1–4 in DMSO-d6 displays a singlet in the range −474.1 to −486.1 ppm (Fig. 11). However, on addition of CT DNA, a downfield shift is observed (in the range −463.1 to −484.4 ppm) with respect to δ value of the complexes in the absence of DNA. The amount of shift of δ value is 3.1, 6.0, 11.0 and 1.7 ppm respectively for 1–4 complexes indicating maximum shift is observed for complex 3, followed by complexes 2, 1 and 4 respectively suggesting the binding ability follows the order: 3 > 2 > 1 > 4. A minor peak is also observed for each of the four complexes in the range −540.8 to −562.4 ppm, which may be due to the complex-cleaved DNA moiety.
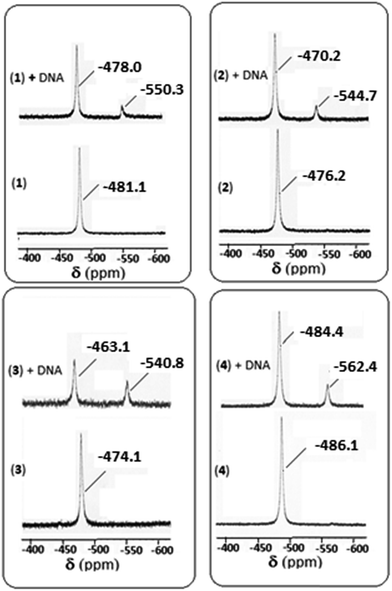 |
| | Fig. 11 51V NMR spectra of complexes 1–4 in absence and in the presence of CT-DNA. | |
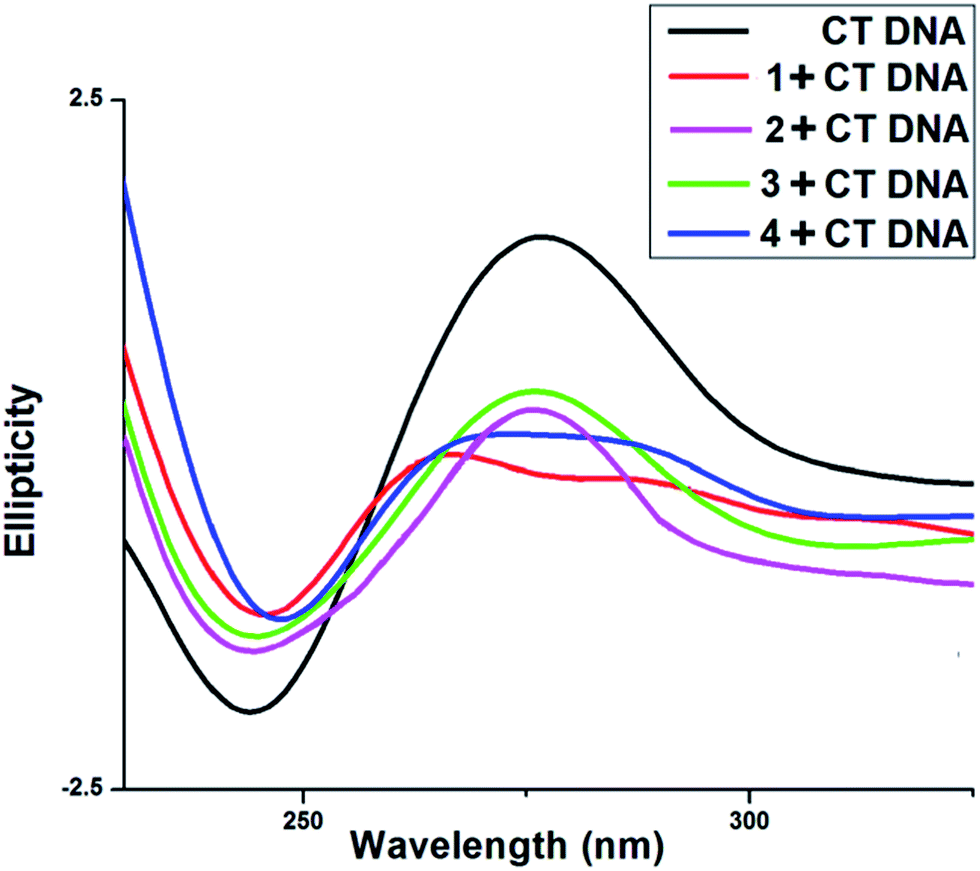
3.6.4. Circular dichroism study. CD spectra of CT DNA shows a positive peak at 275 nm and a negative peak at 245 nm (Fig. 12). The intensity of positive peak at 275 nm along with the negative peak at 245 nm are greatly reduced in the presence of all four vanadium complexes. This positive peak at 275 nm is due to base helicity while the negative peak at 245 nm is due to base stacking interaction. The decrease in the intensity of negative as well as positive bands indicate changes in the DNA double helix conformation. The base stacking and helicity of DNA are slightly altered due to electrostatic interaction and simple groove binding but by contrast intercalation causes with CT DNA that is reflected prominent changes i.e., either increases or decreases in the intensity of both negative and positive bands.46–50,52
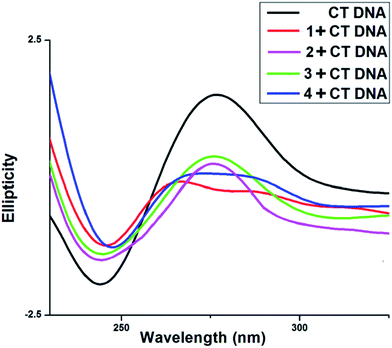 |
| | Fig. 12 Circular dichroism spectra of 200 μL sample containing CT-DNA in 1 mM sodium phosphate buffer (pH 7.2). The scan rate of 50 nm min−1 was maintained and cuvette with 1 mm path length was used. | |
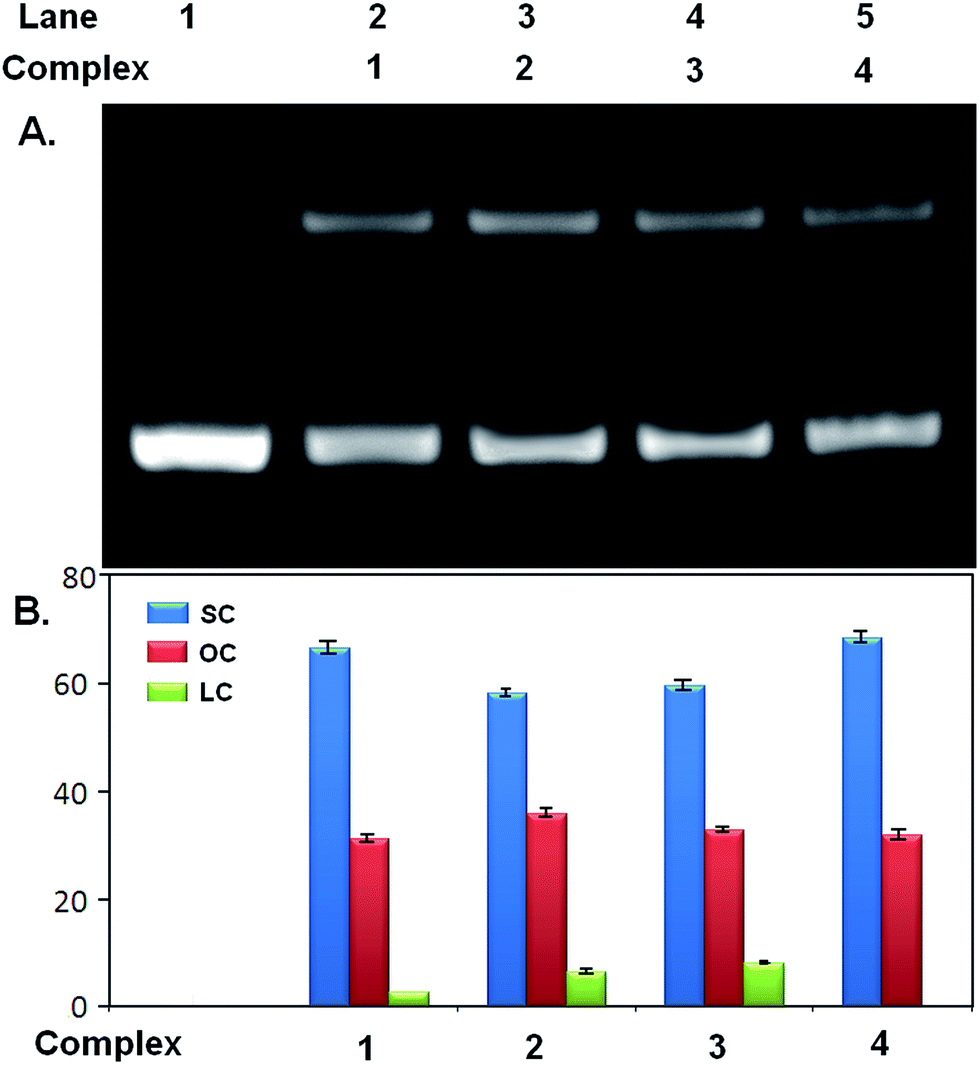
3.7. Nuclease activity of the complexes
The phosphodiester backbone of the DNA accounts for its stability. The relative potential of vanadium complexes to cleave pUC19 plasmid was studied by agarose gel electrophoresis in the absence of any external effect such as UV light, reducing and oxidizing agents. The nuclease efficiency of the complexes was assessed by their ability to convert the supercoiled (SC) form of the DNA to the open circular (OC) relaxed form or the linear coil (LC) form. Relatively faster migration occurs in the case of SC, slower migration for OC and LC migrates in between them. At concentration of 500 μM of the complexes, both linear coil (LC) as well as open circular (OC) form of pUC19 was observed (Fig. 13 and Table 5) whereas at lower concentration (250 μM) only open circular (OC) form was observed (Fig. S9 and Table S7†). The best nuclease activity was shown by complex 3 (possessing –OCH3 group as substituent) i.e., followed up by complex 2 (possessing –CH3), unsubstituted one (complex 1) and complex 4 (possessing –Cl). The –OCH3 group is an electron releasing group that causes the increase in electron density on complex which accounts for enhanced cleavage of the pUC19. The nuclease activity is very poor for complex 4 possessing Cl atom as substituent due to its electron withdrawing nature. So, cleavage efficiency is proportional to the electron releasing ability of the substituents. The vanadium complexes follow the same reactivity order as the DFT optimized η values.53 Here the dominant mechanism responsible for nicking as well as for linearization by the vanadium complexes is the electron transfer from ring substituents followed by hydrolysis of phosphodiester bonds in DNA. The cleaving efficiency of all vanadium complexes has been calculated by their capability to convert supercoiled pUC19 to linear and nicked forms by agarose gel electrophoresis. The vanadium metal is mainly responsible for the cleavage activity of the complex. The ligands used in these complexes were also checked for the nuclease activity. The maximum DNA cleavage by the ligands was observed with H3L3 (ligand of complex 3) at 500 μM which form LC along with SC. The rest three ligands i.e., H3L1, H3L2 and H3L4 form only SC form, the cleaving efficiency does not change with increasing ligand concentration. The ligands H3L1–4 cause the formation of OC form (Fig. S10 and Table S8†). Control experiments suggest that pUC19 DNA alone does not show any DNA cleavage activity under the same experimental conditions.
| % of OC form = (amount of OC form × 100)/[(amount of SC form × 1.42*) + amount of OC form + amount of linear form] |
| (*1.42 = correction factor for supercoiled plasmid) |
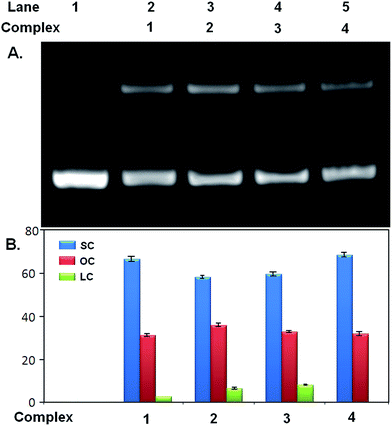 |
| | Fig. 13 Nuclease activity of complexes 1–4. (A) Agarose gel electrophoresis image showing formation of different forms of pUC19 incubated with 0.5 mM of complex 1, 2, 3 and 4. Lane 1 contains pUC19 as control. (B) Quantitative estimation of SC, OC and LC form of pUC19 after incubation with 1–4 from gel image. Error bar values represents mean ± SD. | |
Table 5 Extent of DNA (SC pUC19, 500 ng) cleavage by 0.5 mM 1–4 complexes
| Complex |
pUC form I (% SC) |
pUC form II (% OC) |
pUC form III (% LC) |
| 1 |
66.4 |
24.2 |
2.6 |
| 2 |
57.9 |
28.7 |
5.3 |
| 3 |
59.4 |
26.3 |
9.4 |
| 4 |
68.1 |
24.7 |
0.0 |
3.8. Molecular docking
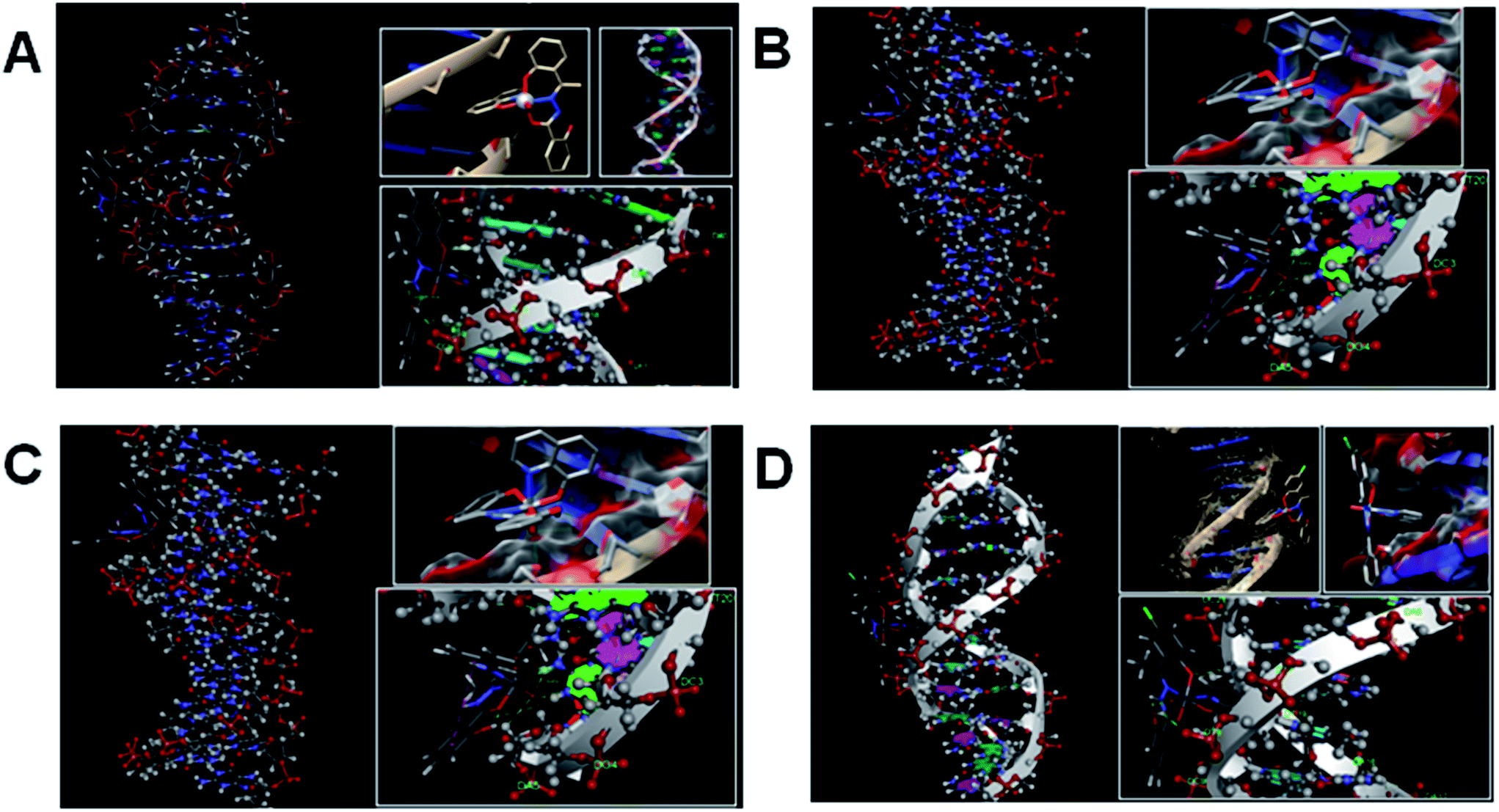
Molecular docking is a very effective technique to determine the binding pattern of small molecules into the binding site of the target specific region of the DNA, mainly in a non-covalent fashion.54 The stability of some protein or DNA intercalated vanadium complex is maintained by van der Waal's forces, hydrogen bonding, stacking interactions and electrostatic and steric effects.55 To the best of our knowledge, we, for the first time have successfully docked four vanadium(V) complexes with the CT-DNA duplex of the sequence d(CGCGAATTCGCG)2 dodecamer (PDB ID: 1BNA) to predict the mode of binding and binding strength of complexes with DNA (Fig. 14). It has been observed that binding of all the drug molecules was stabilized by one or more H-bonds and stacking interaction with DNA bases. The energetically most favourable docking positions were calculated. For complexes 1 and 4 one hydrogen bonding interaction is found between the free phenolic hydroxyl group of complex with a thymine base (A Thy 8) of DNA and three weak π interactions between the phenyl ring of the hydrazone ligand with phosphate and sugar part of DNA whereas for complexes 2 and 3 three hydrogen bonding interactions involving VO and an oxygen atom attached with the oxine molecule and the guanine base (A Gua 4) of DNA and two weak π interactions between the complex and sugar part of DNA (Table S9†). The resulting change of Gibbs free energy of docked structures for complexes 1, 2, 3 and 4 were found to be −7.96, −8.18, −8.35 and −7.89 kcal mol−1 respectively. The values of the Gibbs free energy indicates that the strength of interaction between the complex and DNA follows the order: 3 > 2 > 1 > 4. These energy values for 2 and 3 are in favour of their binding with DNA through intercalation mode and those for 1 and 4 through minor groove binding. Free ligands bind with DNA less effectively than metal complexes (Fig. S11†). The resulting change of Gibbs free energy of docked structures for ligands H3L1, H3L2, H3L3 and H3L4 were found to be −6.72, −7.16, −7.04 and −6.97 kcal mol−1 respectively. The binding ability parameters obtained from spectroscopic studies are compared with the data obtained from molecular docking study in Table 6. Table S9† lists in detail the type of interactions that prevail between the complexes and DNA base which shows that there are three hydrogen bonding interactions between the DNA and each of the complexes 2 and 3 involving vanadyl oxygen and two phenoxy oxygens compared to only one hydrogen bonding interaction with the free OH of the complex and phosphate part of the DNA molecule in 1 and 4.
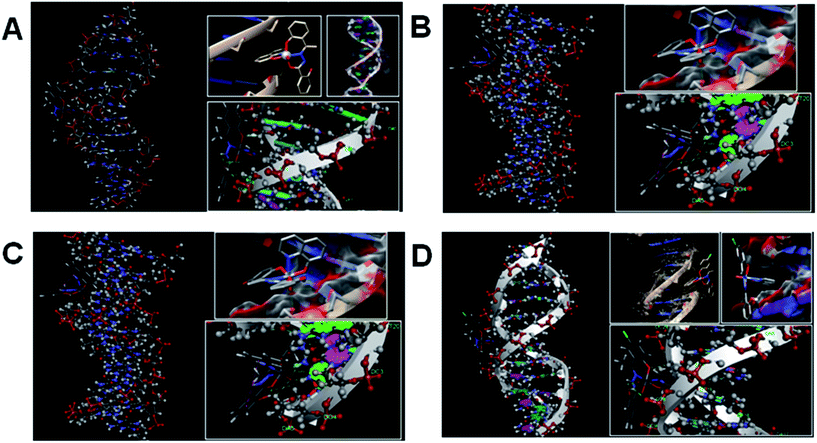 |
| | Fig. 14 Docked pose of complexes ((A) for 1; (B) for 2; (C) for 3 and (D) for 4) showing interaction with DNA base pairs. | |
Table 6 Experimental and computed binding ability parameters of the complexes 1–4 with CT-DNA
| Complex |
UV-spectral study |
Fluorometric study |
Docking |
| Kb (M−1) |
ΔGb (kcal mol−1) |
Kav (M−1) |
ΔGav (kcal mol−1) |
ΔGcal (kcal mol−1) |
| 1 |
3.89 × 105 |
−7.93 |
2.89 × 105 |
−7.74 |
−7.96 |
| 2 |
9.75 × 105 |
−8.49 |
9.75 × 105 |
−8.49 |
−8.18 |
| 3 |
1.65 × 106 |
−8.81 |
1.80 × 106 |
−8.87 |
−8.35 |
| 4 |
1.93 × 105 |
−7.50 |
1.52 × 105 |
−7.35 |
−7.89 |
4. Conclusions
This paper has described the synthesis and a detailed study (viz., structural characterization by X-ray crystallography for all the complexes, solution chemistry, DFT study, DNA binding ability, nuclease activity and molecular docking study) of four novel mixed-ligand oxidovanadium(V) complexes incorporating a family of hydrazone ligands and Hhq as coligand. The molecules reported in this paper form an interesting series because they allow us to examine the electronic effect of substituents in the hydrazone ligands on the chemical as well as physiological properties of the complexes. It has been found that various electronic properties e.g., λmax for the LMCT transition, chemical shift (δ) of the 51V NMR spectra and E½ values of complexes are linearly related with the Hammett parameter (σ) of the substituent. The observed δ values linearly correlate with the inverse energies of the LMCT bands. The electron affinity values of the complexes also correlate with their E½ values. Like the electronic properties, the DNA binding activity as well as nuclease activity also exhibit linear relationship with σ. The experimental bond length and bond angle parameters as well as δ value of 51V NMR spectra have been compared with the theoretical values obtained from DFT studies and are found to be in reasonable agreement. The stability of the complexes was also examined in the DFT study. Molecular docking simulations explored the mode of interaction of the complex molecules with the CT DNA duplex, which to the best of our knowledge, represents the first report of such calculations with mixed-ligand oxidovanadium(V) complexes.
Acknowledgements
We thank University Grants Commission (New Delhi, India) for financial assistance. We are thankful to our College authority for providing research facilities. We gratefully acknowledge DST-FIST for instrumental help at R. K. Mission V. C. College, Rahara. We also thank EPSRC and the University of Reading for funds for the X-Calibur system.
Notes and references
-
(a) D. Patra, N. Biswas, B. Mondal, P. Mitra, M. G. B. Drew and T. Ghosh, RSC Adv., 2014, 4, 22022 RSC;
(b) B. Mondal, M. G. B. Drew and T. Ghosh, Inorg. Chim. Acta, 2010, 363, 2296 CrossRef CAS;
(c) B. Mondal, M. G. B. Drew, R. Banerjee and T. Ghosh, Polyhedron, 2008, 27, 3197 CrossRef CAS;
(d) B. Mondal, T. Ghosh, M. Sutradhar, G. Mukherjee, M. G. B. Drew and T. Ghosh, Polyhedron, 2008, 27, 2193 CrossRef CAS;
(e) T. Ghosh, B. Mondal, T. Ghosh, M. Sutradhar, G. Mukherjee and M. G. B. Drew, Inorg. Chim. Acta, 2007, 360, 1753 CrossRef CAS.
-
(a) D. Rehder, Metallomics, 2015, 7, 730 RSC;
(b) D. Rehder, Met. Ions Life Sci., 2013, 13, 139 Search PubMed;
(c) D. Rehder, Future Med. Chem., 2012, 4, 1823 CrossRef CAS PubMed.
- Z. Wu, Q. Liu, X. Liang, X. Yang, N. Wang, X. Wang, H. Sun, Y. Lu and Z. Guo, J. Biol. Inorg. Chem., 2009, 14, 1413 CrossRef PubMed.
-
(a) E. J. Baran, Adv. Plant Physiol., 2008, 10, 357 CAS;
(b) M. Anke, An. R. Acad. Nac. Farm., 2004, 70, 961 CAS;
(c) H. U. Meisch and H. J. Bielig, Basic Res. Cardiol., 1980, 75, 413 CrossRef CAS PubMed.
-
(a) M. D. Morsy, H. A. Abdel-Razek and O. M. Osman, J. Physiol. Biochem., 2011, 67, 61 CrossRef CAS PubMed;
(b) L. C. Cantley, L. Josephson Jr, R. Warner, M. Yanagisawa, C. Lechene and G. Guidotti, J. Biol. Chem., 1977, 252, 7421 CAS.
-
(a) D. Rehder, G. Santonin, G. M. Licini, C. Schulzke and B. Meier, Coord. Chem. Rev., 2003, 237, 53 CrossRef CAS;
(b) W. Plass, Coord. Chem. Rev., 2003, 237, 205 CrossRef CAS;
(c) K. H. Thompson and C. Orvig, Coord. Chem. Rev., 2001, 219, 1033 CrossRef;
(d) A. Butler, Coord. Chem. Rev., 1999, 187, 17 CrossRef CAS.
- K. H. Thompson, J. H. McNeill and C. Orvig, Chem. Rev., 1999, 99, 2561 CrossRef CAS PubMed.
- K. H. Thompson, B. D. Liboiron, Y. Sun, K. D. D. Bellman, V. Karunaratne, G. Rawji, J. Wheeler, K. Sutton, S. Bhanot, S. B. C. Cassidy, J. H. McNeill, V. G. Yuen and C. Orvig, J. Biol. Inorg. Chem., 2003, 8, 66 CrossRef CAS PubMed.
- H. Yasui, Y. Adachi, A. Katoh and H. Sakurai, J. Biol. Inorg. Chem., 2007, 12, 843 CrossRef CAS PubMed.
- Y. Shechter, I. Goldwaser, M. Mironchik, M. Fridkin and D. Gefel, Coord. Chem. Rev., 2003, 237, 3 CrossRef CAS.
- A. M. B. Bastos, J. G. da Silva, P. I. S. Maia, V. M. Deflon, A. A. Batista, A. V. M. Ferreira, L. M. Botion, E. Niquet and H. Beraldo, Polyhedron, 2008, 27, 1787 CrossRef CAS.
- P. K. Sasmal, A. K. Patra and A. R. Chakravarty, J. Inorg. Biochem., 2008, 102, 1463 CrossRef CAS PubMed.
- I. C. Mendes, L. M. Botion and A. V. M. Ferreira, Inorg. Chim. Acta, 2009, 362, 414 CrossRef CAS.
- J. Benítez, L. Guggeri, I. Tomaz, J. C. Pessoa, V. Moreno, J. Lorenzo, F. X. Avilés, B. Garat and D. Gambino, Z. Anorg. Allg. Chem., 2013, 639, 1417 CrossRef.
- L. H. A. Terra, M. C. Areias, I. Gaubeur and M. E. V. Suez-Iha, Spectrosc. Lett., 1999, 32, 257 CrossRef CAS.
-
(a) Z. Cui, X. Yang, Y. Shi, H. Uzawa, J. Cui, H. Dohi and Y. Nishida, Bioorg. Med. Chem. Lett., 2011, 21, 7193 CrossRef CAS PubMed;
(b) M. R. Maurya, S. Agarwal, M. Abid, A. Azam, C. Bader, M. Ebel and D. Rehder, Dalton Trans., 2006, 937 RSC;
(c) L. Savini, L. Chiasserini, V. Travagli, C. Pellerano and E. Novellino, Eur. J. Med. Chem., 2004, 39, 113 CrossRef CAS PubMed.
-
(a) P. Dandawate, E. Khan, S. Padhye, H. Gaba, S. Sinha, J. Deshpande, S. K. Venkateswara, M. Khetmalas, A. Ahmad and F. H. Sarkar, Bioorg. Med. Chem. Lett., 2012, 22, 3104 CrossRef CAS PubMed;
(b) F. F. Tian, J. H. Li, F. L. Jiang, X. L. Han, C. Xiang, Y. S. Ge, L. L. Li and Y. Liu, RSC Adv., 2012, 2, 501 RSC;
(c) G. S. Hassan, H. H. Kadry, S. M. Abou-Seri, M. M. Ali and A. E. Mahmoud, Bioorg. Med. Chem., 2011, 19, 6808 CrossRef CAS PubMed;
(d) K. Effenberger, S. Breyer and R. Schobert, Eur. J. Med. Chem., 2010, 45, 1947 CrossRef CAS PubMed;
(e) C. D. Fan, H. Su, J. Zhao, B. X. Zhao, S. L. Zhang and J. Y. Miao, Eur. J. Med. Chem., 2010, 45, 1438 CrossRef CAS PubMed;
(f) W. Y. Liu, H. Y. Li, B. X. Zhao, D. S. Shin, S. Lian and J. Y. Miao, Carbohydr. Res., 2009, 344, 1270 CrossRef CAS PubMed;
(g) Y. Xia, C. D. Fan, B. X. Zhao, J. Zhao, D. S. Shin and J. Y. Miao, Eur. J. Med. Chem., 2008, 43, 2347 CrossRef CAS PubMed;
(h) J. Easmon, G. Puerstinger, K. S. Thies, G. Heinisch and J. Hofmann, J. Med. Chem., 2006, 49, 6343 CrossRef CAS PubMed;
(i) T. B. Chaston, R. N. Watts, J. Yuan and D. R. Richardson, Clin. Cancer Res., 2004, 10, 7365 CrossRef CAS PubMed;
(j) A. C. Cunha, J. M. Figueiredo, J. L. M. Tributino, A. L. P. Miranda, H. C. Castro, R. B. Zingali, C. A. M. Fraga, M. C. B. V. Souza, V. F. Ferreira and E. Barreiro, Bioorg. Med. Chem., 2003, 11, 2051 CrossRef CAS PubMed;
(k) L. R. Morgan, B. S. Jursic, C. L. Hooper, D. M. Neumann, K. Thangaraj and B. LeBlanc, Bioorg. Med. Chem. Lett., 2002, 12, 3407 CrossRef CAS PubMed;
(l) G. Darnell and D. R. Richardson, Blood, 1999, 94, 781 CAS;
(m) D. R. Richardson, Antimicrob. Agents Chemother., 1997, 41, 2061 CAS;
(n) G. R. Braslawsky, M. A. Edson, W. Pearce, T. Kaneko and R. S. Greenfield, Cancer Res., 1990, 50, 6608 CAS.
-
(a) J. Benítez, A. C. de Queiroz, I. Correia, M. A. Alves, M. S. Alexandre-Moreira, E. J. Barreiro, L. M. Lima, J. Varela, M. González, H. Cerecetto, V. Moreno, J. C. Pessoa and D. Gambino, Eur. J. Med. Chem., 2013, 62, 20 CrossRef PubMed;
(b) S. P. Dash, S. Pasayat, S. Bhakat, R. Dinda, E. R. T. Tiekink, S. Mukhopadhyay, S. K. Bhutia, M. R. Hardikar, B. N. Joshi, Y. P. Patil and M. Nethaji, Inorg. Chem., 2013, 52, 14096 CrossRef CAS PubMed;
(c) M. R. Maurya, A. A. Khan, A. Azam, S. Ranjan, N. Mondal, A. Kumar, F. Avecilla and J. C. Pessoa, Dalton Trans., 2010, 39, 1345 RSC;
(d) M. R. Maurya, A. A. Khan, A. Azam, A. Kumar, S. Ranjan, N. Mondal and J. C. Pessoa, Eur. J. Inorg. Chem., 2009, 5377 CrossRef CAS.
- B. R. Short, M. A. Vargas, J. C. Thomas, S. O'Hanlon and M. C. Enright, J. Antimicrob. Chemother., 2006, 57, 104 CrossRef CAS PubMed.
- M. Albrecht, M. Fiege and O. Osetska, Coord. Chem. Rev., 2008, 252, 812 CrossRef CAS.
- S. F. Vanparia, T. S. Patel and N. A. Sojitra, Acta Chim. Slov., 2010, 57, 600 Search PubMed.
-
(a) N. Biswas, D. Patra, B. Mondal, M. G. B. Drew and T. Ghosh, J. Coord. Chem., 2015 DOI:10.1080/00958972.2015.1107054;
(b) D. Patra, N. Biswas, B. Mondal, M. G. B. Drew and T. Ghosh, Polyhedron, 2012, 48, 264 CrossRef CAS;
(c) S. Nica, M. Rudolph, H. Görls and W. Plass, Inorg. Chim. Acta, 2007, 360, 1743 CrossRef CAS;
(d) T. Ghosh, S. Bhattacharya, A. Das, G. Mukherjee and M. G. B. Drew, Inorg. Chim. Acta, 2005, 358, 989 CrossRef CAS;
(e) M. R. Maurya, S. Khurana and D. Rehder, Transition Met. Chem., 2003, 28, 511 CrossRef CAS;
(f) S.-X. Liu and S. Gao, Polyhedron, 1998, 17, 81 CrossRef CAS;
(g) J. Chakravarty, S. Dutta, A. Dey and A. Chakravorty, J. Chem. Soc., Dalton Trans., 1994, 557 RSC.
-
(a) M. Alagesan, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2013, 42, 7210 RSC;
(b) P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2012, 41, 4423 RSC;
(c) D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, J. Biol. Inorg. Chem., 2012, 17, 223 CrossRef CAS PubMed;
(d) P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376 CrossRef CAS PubMed;
(e) Z. C. Liu, B. D. Wang, B. Li, Q. Wang, Z. Y. Yang, T. R. Li and Y. Li, Eur. J. Med. Chem., 2010, 45, 5353 CrossRef CAS PubMed;
(f) K. Ghosh, P. Kumar, N. Tyagi, U. P. Singh, V. Aggarwal and M. C. Baratto, Eur. J. Med. Chem., 2010, 45, 3770 CrossRef CAS PubMed;
(g) Q. Wang, Z. Y. Yang, G. F. Qi and D. D. Qin, Eur. J. Med. Chem., 2009, 44, 2425 CrossRef CAS PubMed;
(h) Z. C. Liu, B. D. Wang, Z. Y. Yang, Y. Li, D. D. Qin and T. R. Li, Eur. J. Med. Chem., 2009, 44, 4477 CrossRef CAS PubMed;
(i) Y. Li, Z. Y. Yang and M. F. Wang, Eur. J. Med. Chem., 2009, 44, 4585 CrossRef CAS PubMed;
(j) T. R. Li, Z. Y. Yang, B. D. Wang and D. D. Qin, Eur. J. Med. Chem., 2008, 43, 1688 CrossRef CAS PubMed;
(k) B. D. Wang, Z. Y. Yang, P. Crewdson and D. Q. Wang, J. Inorg. Biochem., 2007, 101, 1492 CrossRef CAS PubMed;
(l) Z. Y. Yang, B. D. Wang and Y. H. Li, J. Organomet. Chem., 2006, 691, 4159 CrossRef CAS;
(m) B. D. Wang, Z. Y. Yang and T. R. Li, Bioorg. Med. Chem., 2006, 14, 6012 CrossRef CAS PubMed;
(n) T. B. Chaston and D. R. Richardson, J. Biol. Inorg. Chem., 2003, 8, 427 CAS.
- R. A. Rowe and M. M. Jones, Inorg. Synth., 1957, 5, 113 CrossRef CAS.
- S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, Y. P. Patil, M. Nethaji, W. Kaminsky, S. Mukhopadhyay and S. K. Bhutia, Dalton Trans., 2014, 43, 10139 RSC.
- Bruker, SAINT (Version 6.36a) Bruker AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- CrysAlis, Oxford Diffraction Ltd, Abingdon, U. K., 2006 Search PubMed.
- Bruker, SMART (Version 5.625) and SADABS (Version 2.03a) Bruker AXS Inc, Madison, Wisconsin, USA, 2001 Search PubMed.
- Abspack, Oxford Diffraction Ltd., U. K., 2005 Search PubMed.
- G. M. Sheldrick, SHELXS97 and SHELXL97: Programs for Crystal Structure Solution and Refinement, University of Göttingen, Germany, 1997 Search PubMed.
-
(a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS;
(b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS;
(c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
-
(a) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS;
(b) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
-
(a) V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS;
(b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS;
(c) P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209 CrossRef CAS;
(d) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS;
(e) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, (Revision A.1), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
-
(a) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS;
(b) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS;
(c) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Below, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL molecular graphics system, DeLano Scientific, San Carlos, CA, USA, 2002 Search PubMed.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376 CrossRef CAS PubMed.
- M. R. Efink and C. A. Ghiron, Anal. Biochem., 1981, 114, 199 CrossRef.
-
(a) M. R. Maurya and N. Kumar, J. Mol. Catal. A: Chem., 2014, 383–384, 172 CrossRef CAS;
(b) M. R. Maurya, C. Haldar, A. Kumar, M. L. Kuznetsov, F. Avecilla and J. C. Pessoa, Dalton Trans., 2013, 42, 11941 RSC;
(c) M. R. Maurya, S. Khurana, C. Schulzke and D. Rehder, Eur. J. Inorg. Chem., 2001, 779 CrossRef CAS;
(d) S. P. Perlepes, D. Nicholls and M. R. Harrison, Inorg. Chim. Acta, 1985, 102, 137 CrossRef CAS.
-
(a) C. R. Cornman, G. J. Colpas, J. D. Hoeschele, J. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1992, 114, 9925 CrossRef CAS;
(b) D. Rehder, C. Weidemann, A. Duch and W. Priebsch, Inorg. Chem., 1988, 27, 584 CrossRef CAS.
- S. Nica, A. Buchholz, M. Rudolph, A. Schweitzer, M. Wächtler, H. Breitzke, G. Buntkowsky and W. Plass, Eur. J. Inorg. Chem., 2008, 2350 CrossRef CAS.
- R. G. Pearson, Hard and Soft acids and bases, Dowden, Hutchinson, Ross, Stroudsburg, PA, 1973 Search PubMed.
- R. G. Pearson, J. Chem. Educ., 1987, 64, 561 CrossRef CAS.
-
(a) U. Saha and K. K. Mukherjea, Int. J. Biol. Macromol., 2014, 66, 166 CrossRef CAS PubMed;
(b) S. Patra, S. Chatterjee, T. K. Si and K. K. Mukherjea, Dalton Trans., 2013, 42, 13425 RSC;
(c) N. H. Khan, N. Pandya, N. C. Maity, M. Kumar, R. M. Patel, R. I. Kureshy, S. H. R. Abdi, S. Mishra, S. Das and H. C. Bajaj, Eur. J. Med. Chem., 2011, 46, 5074 CrossRef CAS PubMed.
- T. K. Si, S. S. Paul, M. G. B. Drew and K. K. Mukherjea, Dalton Trans., 2012, 41, 5805 RSC.
- P. K. Sasmal, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Dalton Trans., 2010, 39, 7104 RSC.
- R. Eshkourfu, B. Čobeljić, M. Vujčić, I. Turel, A. Pevec, K. Sepčić, M. Zec, S. Radulović, T. Srdić-Radić, D. Mitić, K. Andjelković and D. Sladić, J. Inorg. Biochem., 2011, 105, 1196 CrossRef CAS PubMed.
-
(a) S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, Y. P. Patil, M. Nethaji, W. Kaminsky, S. Mukhopadhyay, S. K. Bhutia, W. Kaminsky and E. Sinn, RSC Adv., 2015, 5, 51852 RSC;
(b) S. P. Dash, A. K. Panda, S. Pasayat, S. Majumder, A. Biswas, W. Kaminsky, S. Mukhopadhyay, S. K. Bhutia and R. Dinda, J. Inorg. Biochem., 2015, 144, 1 CrossRef CAS PubMed.
-
(a) F. Arjmand and S. Parveen, RSC Adv., 2012, 2, 6354 RSC;
(b) G. Zhang, J. Guo, J. Pan, X. Chen and J. Wang, J. Mol. Struct., 2009, 923, 114 CrossRef CAS.
- M. Ebel and D. Rehder, Inorg. Chem., 2006, 45, 7083 CrossRef CAS PubMed.
- D. Das and P. Mondal, New J. Chem., 2015, 39, 2515 RSC.
-
(a) E. P. Irgi, G. D. Geromichalos, S. Balala, J. Kljun, S. Kalogiannis, A. Papadopoulos, I. Turel and G. Psomas, RSC Adv., 2015, 5, 36353 RSC;
(b) K. Suntharalingam, O. Mendoza, A. A. Duarte, D. J. Mann and R. Vilar, Metallomics, 2013, 5, 514 RSC;
(c) S. Tabassum, W. M. Al-Asbahy, M. Afzal, F. Arjmand and V. Bagchi, Dalton Trans., 2012, 41, 4955 RSC;
(d) Y. Y. Fang, B. D. Ray, C. A. Claussen, K. B. Lipkowitz and E. C. Long, J. Am. Chem. Soc., 2004, 126, 5403 CrossRef CAS PubMed.
-
(a) C. Datta, D. Das, P. Mondal, B. Chakraborty, M. Sengupta and C. R. Bhattacharjee, Eur. J. Med. Chem., 2015, 97, 214 CrossRef CAS PubMed;
(b) Z. L. You, H. Sun, B. W. Ding, Y. P. Ma, M. Zhang and D. M. Xian, J. Coord. Chem., 2011, 64, 3510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP view of the H3L3 ligand; cyclic voltammogram at different scan rate; fluorescence spectra of the ligands; DNA cleavage activity of the ligands; docked pose of the ligands; Fig. S1–S11; Tables S1–S9. CCDC 1406044–1406048 contains the supplementary crystallographic data for H3L3 and 1–4 complexes. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra17844d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.