
Fabrication of conductive oxidase-entrapping nanocomposite of mesoporous ceria–carbon for efficient electrochemical biosensor†
Abstract
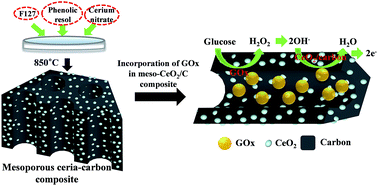
A conductive nanocomposite containing an immobilized oxidative enzyme in the pores of mesostructured ceria (CeO2)–carbon was developed as an efficient electrochemical biosensing platform. The construction of the nanocomposite began with the incorporation of CeO2 in a carbon matrix by the co-assembly of cerium nitrate, resol, and triblock copolymer via a facile evaporation-induced self-assembly method, which resulted in the formation of mesoporous ceria–carbon (denoted as Meso-CeO2/C). Glucose oxidase (GOx) was subsequently immobilized in the vacant pores of the Meso-CeO2/C by using glutaraldehyde crosslinking to prevent enzyme leaching from the matrix. H2O2 generated by the catalytic action of GOx in proportion to the amount of target glucose was rapidly converted into hydroxyl radicals by the catalytic activity of CeO2, which induced subsequent anodic oxidation of Ce3+ into Ce(OH)22+ or Ce(OH)4 with the anodic current. The constructed Meso-CeO2/C exhibited higher resolution in electrochemical detection of H2O2 than pure mesoporous carbon without ceria owing to the catalytic activity of ceria. The anodic current responses by the nanocomposite containing GOx in Meso-CeO2/C resulted in a linear increase in the concentration of target glucose (0.25–5 mM), which is suitable to measure the serum glucose, with excellent storage stability of over two months at room temperature. The biosensor also exhibited a high degree of precision and reproducibility when employing real human blood samples. Based on these results, we anticipate that this novel biosensing format can be readily extended to other oxidative enzymes for the detection of various clinically important target molecules.
 Please wait while we load your content...
Please wait while we load your content...

