
Constructing pendent imidazolium-based poly(phenylene oxide)s for anion exchange membranes using a click reaction†
Abstract
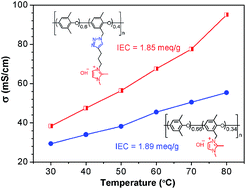
A series of poly(phenylene oxide)s (PPOs) bearing a flexible pendent imidazolium cation were prepared by an azide–alkyne cycloaddition between azidomethylated PPO and a novel alkyne-containing imidazolium, and their structures were confirmed by 1H NMR, 13C NMR, and FT-IR. The corresponding anion exchange membranes (AEMs) showed distinct hydrophobic/hydrophilic phase-separated morphology at higher imidazolium content, as evidenced by AFM and SAXS techniques, which favors for the construction of interconnected hydroxide transport channels. As a result, the as-prepared AEMs exhibited higher conductivity (95 mS cm−1, 80 °C, 100% RH) than conventional imidazolium benzylic-type AEM (55 mS cm−1, 80 °C, 100% RH) with even lower IEC. Furthermore, the introduction of a 1,2,3-triazole moiety into the polymer side chain does not compromise its thermal and alkaline stability. This investigation demonstrated that the “click chemistry” strategy will benefit further tailoring of high performance AEMs with “side-chain-type” architectures.
 Please wait while we load your content...
Please wait while we load your content...

