
Visible-light-driven dye degradation using a floriated ZnIn2S4/AgIn5S8 heteromicrosphere catalyst†
Jiangluqi Songa,
Tongtong Jianga,
Gengwu Jib,
Wenting Zhanga,
Xiangyi Chenga,
Wei Wenga,
Lixin Zhu*c and
Xiaoliang Xu*a
aKey Laboratory of Strongly-Coupled Quantum Matter Physics, Chinese Academy of Sciences, and Department of Physics, University of Science and Technology of China, Hefei, Anhui 230026, China
bShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201204, China
cCenter Laboratory, First Affiliated Hospital of Anhui Medical University, Hefei 230026, China
First published on 23rd October 2015
Abstract
We report the synthesis of ZnIn2S4/AgIn5S8 (ZIS/AIS) heteromicrospheres with tailorable composition and controllable band gap via partial cation exchange for the first time. Detailed studies of the as-prepared samples were carried out using SEM, TEM, STEM-EDS, XRD and layer-by-layer depth XPS analysis. Based on these results, possible cation exchange was proposed, and a critical concentration of Ag+ in the crystals was identified, below which cation exchange cannot occur. Besides, an enhanced photodegradation mechanism was discussed by analyzing the UV-vis-NIR reflectance spectra and the band structure, illustrating that an appropriate thickness of the AIS layer is beneficial to the generation of active species thus favoring the degradation reaction. Further application of dye degradation demonstrated a remarkable photodegradation rate up to 99.2%. Our results shed light on how the cation exchange occurs between ternary and binary crystals, which may open a new avenue to the field for synthesizing materials at the micro-scale.
1. Introduction
Ternary semiconductors, such as Ag–In–S (AIS) and Cu–In–S, containing a “non-Class A metal”, have gained intensive attention in biolabeling, opto-electronic devices and solar cells for their wonderful biocompatibility, good defect tolerance and excellent optical properties.1–10 Among them, ZnIn2S4 (ZIS) semiconductors, have attracted considerable interest in light emitting diodes and photocatalysis because of their lower cost and more convenient approach than that of I–III–VI materials.1,7,11–21 However, ZIS semiconductors did not exhibit an admirable light utilization efficiency compared to AIS (band gap 1.8–2.1 eV) material in photocatalysis and solar cells because of their relatively wide band gap (2.4–2.8 eV).22,23 Apparently, exploration of new strategies for producing materials with the combined advantages of these two kinds of semiconductors is still expected.In the field of material synthesis, meanwhile, of particular interest is the cation exchange method which can preserve the pristine crystal frameworks, playing a significant role in producing materials of diverse shapes and desired compositions.1,4,24–31 Giving the obvious merits, a series of achievements have been made during recent years. Son and his co-workers30 found that the shapes of obtained nanocrystals were pronouncedly influenced by their critical size during the cation exchange process. Akkerman et al.20 realized the transformation from binary nanocrystals to ternary and quaternary nanocrystals by adjusting the exchange sequence. Miszta et al.27 presented an example of a nanostructure in which the branching structure was achieved by the appropriate organization of different domains in a single nanocrystal. van der Stam’s32 group has prepared luminescent CuInS2 quantum dots starting from Cu–S binary nanocrystals. Yet, no research of the cation exchange reaction at the micro-scale in an aqueous system has been reported. In addition, there still exists a crucial question of how exactly the cations in the initial crystal are replaced by the guest cations.
In this paper, cubic ZnIn2S4/AgIn5S8 (ZIS/AIS) hetero-microspheres (HMSs) with visible light absorption were successfully synthesized via partial cation exchange. A possible mechanism for the cation exchange was proposed on the basis of XRD and XPS depth measurements, according to which pure AIS and Ag2S MSs were also prepared. By analyzing the optical absorption, the band structure of ZIS/AIS HMSs was obtained and the mechanism for enhanced photodegradation was discussed. What is more, a visible light driven dye degradation test was performed, showing a high photodegradation rate up to 99.2%. Our work demonstrates that, in place of a doping method, cation exchange could be used as a facile and versatile approach for synthesis of a band controllable and composition tailorable ZIS/AIS heterostructure which has potential applications in solar cells and for hydrogen evolution.
2. Experimental section
2.1 Chemicals
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99.99%), silver nitrate (AgNO3, 99.99%), thioacetamide (TAA, 99%), nitric acid, methanol, and absolute ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Indium acetate (In(OAc)3, 99.99%) was purchased from Alfa Aesar Chemical Reagent Co. (China). All chemicals were of analytical grade and used as received without further purification. All the water used in experiments was deoxidized and deionized (18.25 MΩ cm).2.2 Synthesis of floriated ZIS microspheres
Floriated ZIS microspheres were synthesized via a hydrothermal process according to the previous report with large modifications.15,33 Typically, 2 mmol of Zn(NO3)2·6H2O, 4 mmol of In(OAc)3·4.5H2O, and 10 mmol of thioacetamide (TAA) were dissolved in 30 mL of water. Then, 1 M nitric acid was introduced so that pH = 2 was achieved. Subsequently, the mixture was sealed in a Teflon-lined stainless steel autoclave, and heated to 90 °C with a heating rate of 5 °C min−1 and was kept at this temperature for 4 h. After naturally cooling to room temperature, the orange precipitates were washed with water and ethanol alternately and dried at 70 °C for further use. The Ag–In–S microspheres were synthesized similarly except that the amount of AgNO3 was 1 mmol.2.3 In situ cation exchange preparation of ZIS/AIS heteromicrospheres (HMSs)
A certain amount of AgNO3 and 0.001 mmol of In(OAc)3·4.5H2O were dissolved in 10 mL water, and added into 20 mL of water which contained 1 mmol of previously synthesized ZIS MSs and 800 μL of methanol. Then the mixture was transferred into an autoclave and kept at 90 °C for 3 h. Then the temperature was raised to 150 °C and was kept at this temperature for another 3 h. The purification and drying procedures of the ZIS/AIS HSMSs were the same as that of the ZIS MSs. In this work, we set the molar ratio of Ag to Zn (total Zn in the ZIS MSs) as 0.5, 0.75, 1 and 1.25, noting as H1–H4.2.4 Material characterization
The transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) images were measured using a JEOL JEM 1400 working at 200 kV. The elemental line profiles were performed using a JEOL JEM-2100F equipped with a field emission gun working at 200 kV. The general morphology of the samples was investigated using a scanning electron microscopy (SEM, JEOL JSM 6700F) system equipped with an energy dispersive X-ray spectrometry (EDX) attachment. The elemental content of the samples was detected using a Shimadzu X-ray fluorescence (XRF) spectrometer (XRF 1800). XRD measurements were conducted on a MXPAHF 18 kW diffractometer with Cu Kα (0.154 nm) serving as the incident radiation. The samples were pre-prepared by thorough grinding and drying. The UV-vis diffuse reflectance spectra (DRS) were acquired by a Shimadzu Solid 3700 UV-vis-NIR spectrophotometer equipped with an integrating sphere. The Brunauer–Emmett–Teller (BET) surface area was measured with ASAP2010 M apparatus. All the samples were degassed at 120 °C overnight prior to BET measurements. The nitrogen adsorption and desorption isotherms were measured at −196 °C.2.5 X-ray photoelectron spectroscopy (XPS) depth profiles
XPS profiles were measured using a Phoibos 100 analyzer with a pass energy of 10 eV which was calibrated according to the Fermi level (EF) of Au. Standard Al Kα (1486.61 eV) composed of a monochromator was used as the X-ray source with a base ultrahigh vacuum of 10−10 mbar. The resolution of XPS was 0.6 eV measured as the full width at half maximum (FWHM) of the Ag 3d5/2 peak with a pass energy of 10 eV. The depth profile analyses were performed by bombarding the surface of the samples with Ar+ ions accelerated at a high voltage of 1.5 kV with a sample current of 1.5 μA. The sputtering rate of the samples was about 1 nm min−1 under these pre-set conditions, and data were collected at an interval of 5 min. All the spectra were corrected by the C 1s peak at 285.0 eV. Quantitative elemental compositions were measured from peak areas using experimentally determined sensitivity factors and the spectrometer transmission function. The high-resolution spectra were deconvoluted to the Gaussian–Lorentzian function by means of the Casaxps software.2.6 Photocatalytic degradation of dye
Rhodamine B (RhB) was used to evaluate the photodegradation performance of the prepared MSs and HMSs. The standard absorption curve was calibrated by measuring the absorbance of various concentrations of Rhodamine B solution at 554 nm. 30 mg of the photocatalyst, i.e. ZIS MSS and ZIS/AIS HMSs (sample H1–H4), was respectively suspended in 100 mL of a solution containing 60 ppm of RhB. Prior to irradiation, the suspension was magnetically stirred in a dark environment to ensure the establishment of an adsorption–desorption equilibrium. At a given interval, a small amount of the suspension was taken out, centrifuged, and the supernatant was analysed using a UV-vis spectrophotometer. After the pre-adsorption (PA), a 500 W tungsten halogen lamp equipped with a cutoff filter that can remove wavelengths less than 420 nm served as the visible light source. To monitor the degradation, aliquots of the sample were taken out at different time intervals to examine their absorption spectra (as described above). The rate of photodegradation was calculated using the equation, R = (C0 − C) × 100%/C0, where R, C0 and C represent the degradation rate, initial concentration and the current concentration of different intervals, respectively.3. Results and discussion
3.1 Morphology and crystal structure
Fig. 1a is the representative SEM image of the initial ZIS MSs. It shows that the product is a solid sphere, which can be deduced from some broken parts (shown in Fig. S1†), just like a peony, with an average diameter of 5 μm. The surface is composed of plenty of nanosheets/nanoflakes, which is more homogeneous than previously reported.33 The flaky mesoporous structure may be ideal for photocatalytic reaction because of the large specific surface area. Fig. 1c–f are the SEM images of ZIS/AIS HMSs prepared in situ by cation exchange reaction under the same experimental conditions but with different molar ratios of the reaction precursors (Ag to ZIS) of 0.5, 0.75, 1.1 and 1.25, noted as samples H1–H4. They reveal that, in contrast to the doping method15 and compared with a directly synthesized AIS catalyst using a hydrothermal protocol (Fig. S2†), the HMSs respectably preserved the flake-like and porous structure, except for some slight crimps on the edge of the nanosheets (Fig. 1d and e). This may be the result of a distortion caused by the mismatch between the AIS and ZIS crystals in the surface region of the nanosheets. Lots of nanosheets are clearly seen on the surface of the MSs from the TEM image (Fig. 1f). Fig. 1g is the HRTEM image of a nanosheet belonging to a pristine ZIS microsphere, showing the (211) and (310) lattice sets of cubic ZnIn2S4 with interplanar spacings of 4.35 and 3.36 Å, respectively. Besides, the HRTEM images of H1–H4 also show (310), (220) and (202) lattice sets of cubic AgIn5S8, indicating the formation of AIS components. Normalized by the intensity of the S peak, the EDX spectrum (Fig. 1l) shows the characteristic peaks of Zn Lα, In Lα and S Kα of ZIS MSs with an intensity of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4, consistent with the stoichiometric form of ZnIn2S4, and the Ag Lα peak of the ZIS/AIS HMSs. The magnified spectra of Zn Lα and Ag Lα shown in Fig. 1m and n demonstrate that there is a decrease of Zn content and an emergence of Ag content respectively, which qualitatively clarified the reaction of cation exchange of Zn2+ to Ag+.
:2:4, consistent with the stoichiometric form of ZnIn2S4, and the Ag Lα peak of the ZIS/AIS HMSs. The magnified spectra of Zn Lα and Ag Lα shown in Fig. 1m and n demonstrate that there is a decrease of Zn content and an emergence of Ag content respectively, which qualitatively clarified the reaction of cation exchange of Zn2+ to Ag+.
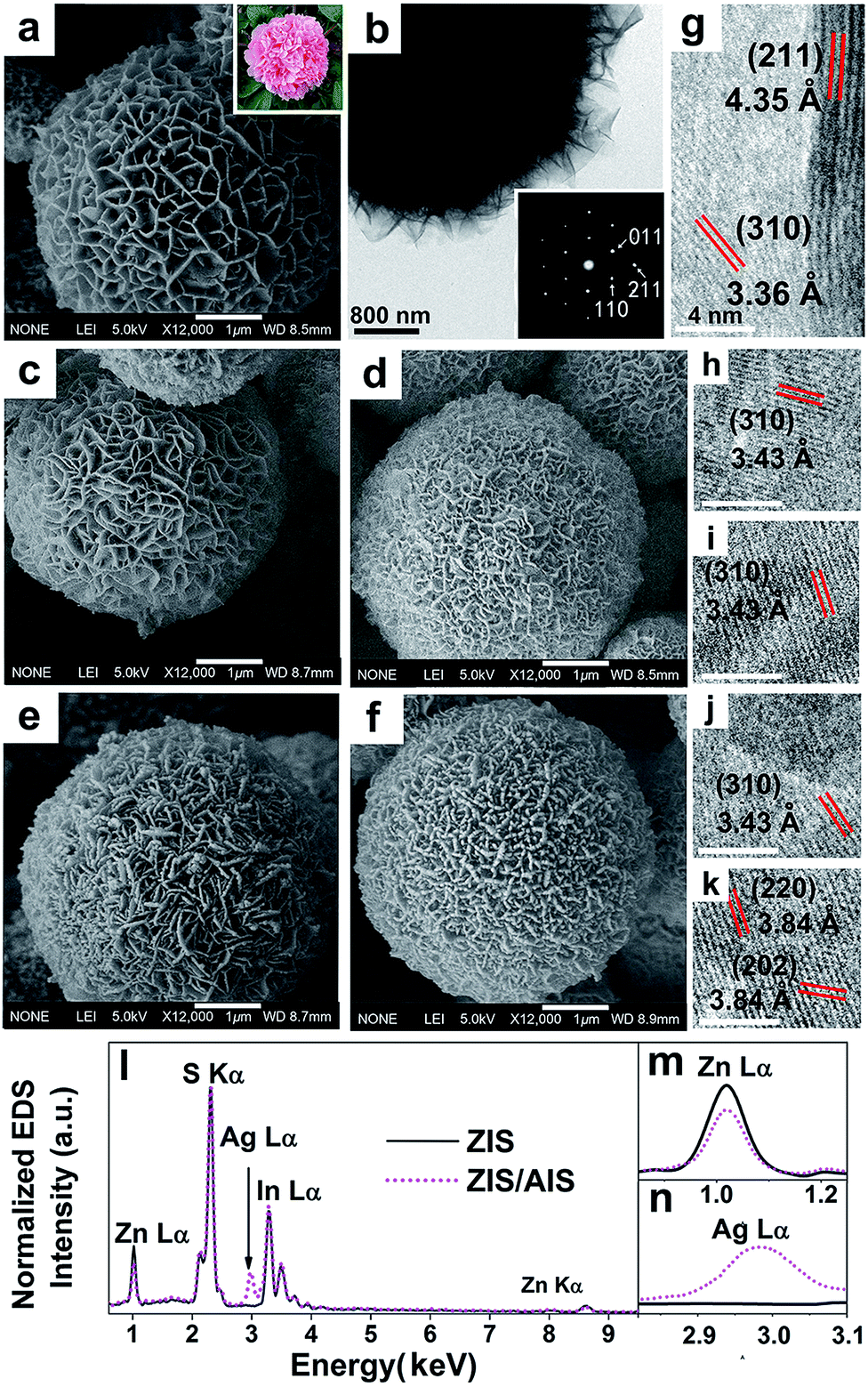 | ||
| Fig. 1 SEM images of (a) initial ZIS MSs and (c–f) the HMSs for samples H1–H4 (inset of (a): photograph of a real peony flower). (b) TEM image of initial ZIS MSs (inset: corresponding Fourier transform). (g–k) HRTEM characterization of the aforementioned samples. The EDS of the (l) pristine ZIS MSs and ZIS/AIS HMSs H1, and their evolution of (m) Zn Lα and (n) Ag Lα spectra (scale bar: 4 nm). | ||
It is noteworthy that the In content seems to remain relatively invariant, which was also seen in other cation reactions of Zn to AgInS2 (ref. 4) and Zn to CuInS2.34 Confirmed by XRF measurement as shown in Table 1, there is a continuous decrease of Zn content accompanied by an increase of Ag, while the In content stays nearly constant. Because of the smaller radius of Zn2+ (0.74 Å) which suffers from a weaker potential barrier during migration than In3+ (0.81 Å) in a lattice,5,35 it is conceivable that Zn2+ has priority to exchange with Ag+. However, we consider this is not believable enough. If this is the case, Ag+ which has a larger radius (1.26 Å) can hardly enter the ZIS lattice,36,37 let alone be able to exchange with Zn2+. A more likely explanation arises when we considered the configuration of atoms in cubic X–In–S (X stands for Ag, Cu or Zn). The unit cell structure diagram of cubic ZnIn2S4 shows (Scheme 1) that each In atom is bonded to the six nearest S atoms, yet there are four S atoms around a Zn atom. As a result, In possesses a more stable chemical state than Zn, and they have more difficulty exchanging with surrounding cations, such as Ag+, Cu+ and Zn2+.
| Sample | Zn/wt% | Ag/wt% | In/wt% | S/wt% | Composition |
|---|---|---|---|---|---|
| ZIS | 14.74 | — | 55.06 | 30.20 | ZnIn2.13S4.19 |
| H1 | 13.03 | 2.64 | 54.46 | 29.86 | Zn0.9Ag0.11In2.13S4.19 |
| H2 | 11.90 | 5.00 | 53.70 | 29.44 | Zn0.83Ag0.21In2.12S4.17 |
| H3 | 11.00 | 6.64 | 52.97 | 29.39 | Zn0.77Ag0.28In2.10S4.18 |
| H4 | 9.91 | 7.87 | 52.79 | 29.43 | Zn0.69Ag0.33In2.08S4.16 |
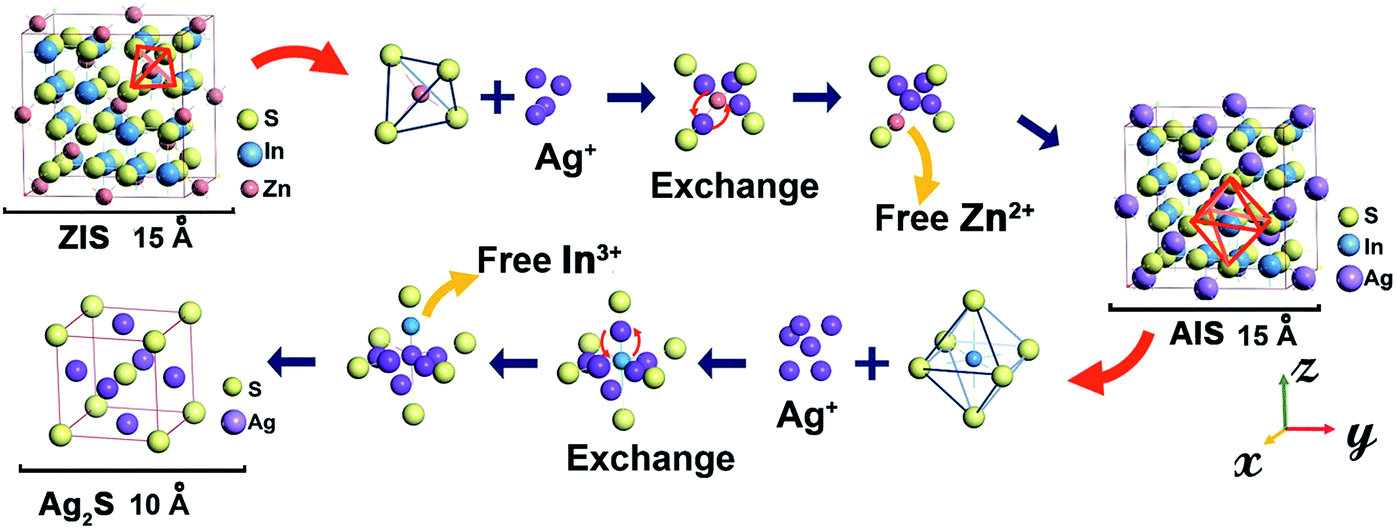 | ||
| Scheme 1 Schematic diagram of cation exchange mechanism from ZIS to Ag2S. | ||
The phase and crystallographic structure of the products were determined using XRD. Fig. 2 shows that all the diffraction peaks of the initial ZIS MSs could be indexed to a cubic phase of ZnIn2S4 (JCPDS no. 48-1778, a = 10.621 Å),11 and no other impurities such as In2S3 or ZnS are found. Similarly, the profiles of the ZIS/AIS HMSs samples are the same as the cubic ZnIn2S4, except for the appearance of (422) and (620) diffraction peaks of cubic AgIn5S8 (JCPDS no. 25-1329, a = 10.825 Å).34 It doesn’t seem strange that all the XRD patterns exhibit the same crystalline phase, for the reason that the anion sublattice, i.e. S2− subframes, maintained their structural rigidity and shape during the exchange reaction.23,28,35 Because of the similar ternary composition, the same bonding situation and the identical atomic sites between AgIn5S8 and ZnIn2S4, the positions of the diffraction peaks hold almost the same lines. Nevertheless, the interplanar spacing (a = 10.825 Å of cubic AgIn5S8) has a tiny distinction on account of the different ion radii between Zn2+ and Ag+ which resulted in a shift of the peaks. So as a consequence, there is a broadening of the (311) and (440) diffraction peaks in the profiles of samples H2–H4. Due to the relatively large size of the HMSs and the uneven surface, the STEM-EDS curves don’t show a clear and accurate elemental distribution of the as-prepared HMSs, except for a roughly estimated thickness of 70 nm for the AIS region (sample H1) (shown in ESI Fig. S3†).
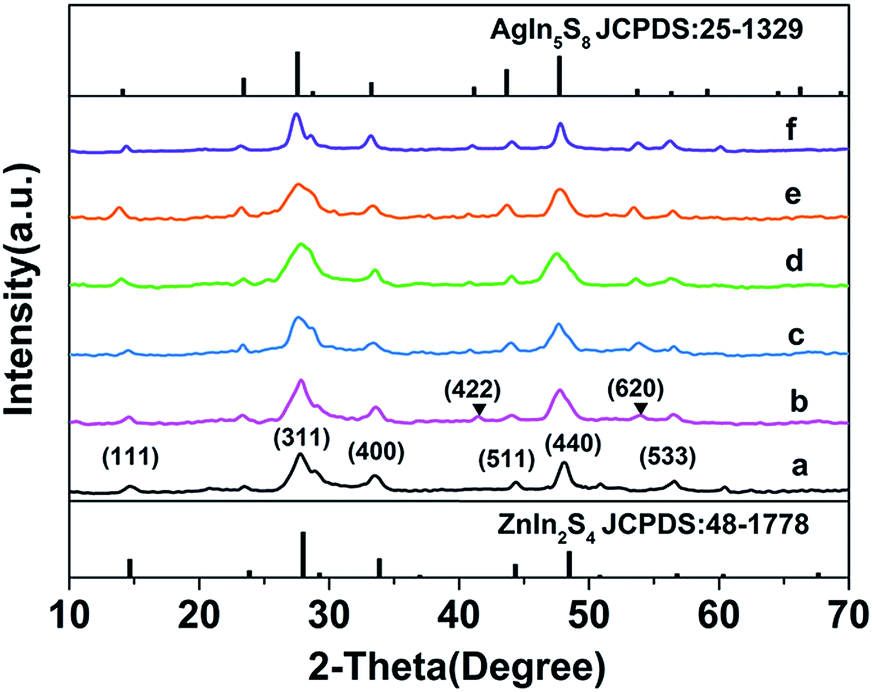 | ||
| Fig. 2 XRD patterns of pristine ZIS MSs (a), samples H1–H4 (b–e) and pure AgIn5S8 (f) derived from cation exchange (▼ indicates the peaks of the cubic AIS phase). | ||
3.2 Influence of experimental conditions
ZIS MSs were synthesized by template-free hydrothermal protocols in the absence of templates or surface ligands. According to the acid–base theory, as a soft base, S2− can easily react with Zn2+ and In3+, which are Lewis bases, to form a stable product, and self-assemble to a microsphere with irregular nanosheets. A previous study has shown that the reaction temperature and pH value have a significant effect on the size and morphology of ZIS MSs.10 Here in this paper, as the ZIS/AIS HMSs were prepared via a cation exchange method for the first time, we only focus on the influence of composition on the reaction temperature and the concentration of the Ag+ precursor in sample H1 during the exchange process, where we fixed all the other experimental variables as detailed in the Experimental section.exp(−ΔHFrenkel/2kT) (ΔHFrenkel is the formation enthalpy of the Frenkel defect), increased accordingly and thus the amount of exchange cations augmented.
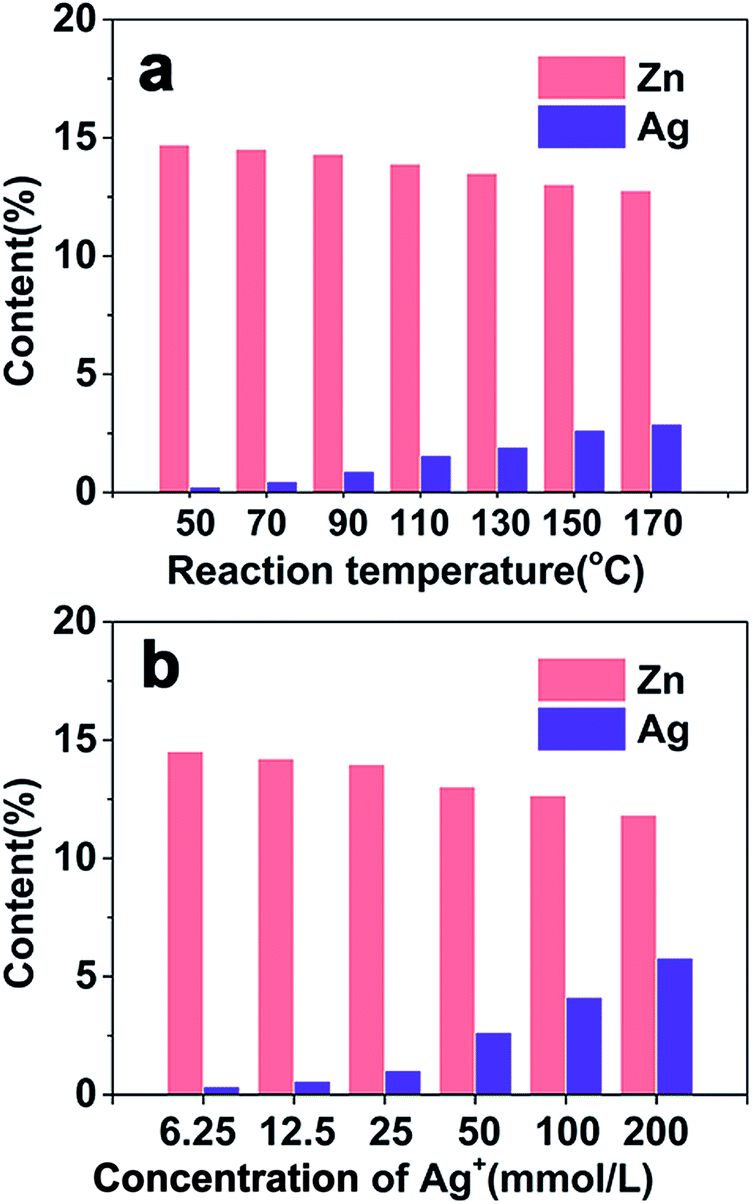 | ||
| Fig. 3 Dependence of the Zn and Ag content on different (a) reaction temperatures and (b) concentrations of Ag+. | ||
During the experiment, we noticed that a rapid color change (from orange to brown, ≪1 s) of the ZIS MS dispersion was observed upon adding an excess amount of AgNO3 without the addition of In(OAc)3 and heating. We investigated the XRD patterns of the samples before and after the addition of the Ag precursor (shown in Fig. 4). Not unexpectedly, two weak but evident diffraction peaks, which can be indexed to the (200) and (220) crystal faces of cubic Ag2S (JCPDS no. 04-0774, a = 4.89 Å), are observed. This means that there was a tiny amount (about several layers) of Ag2S formed on the very edge of the ZIS MSs and the cation exchange between Ag+ and In3+ occurred, otherwise the X-ray diffraction phenomena wouldn’t happened and the characteristic diffraction peaks wouldn’t appear during the XRD measurement. This raises a question regarding how the cation exchange process reacts, or whether the concentration of Ag+ has an effect upon the aqueous exchange reaction in ternary semiconductor materials.
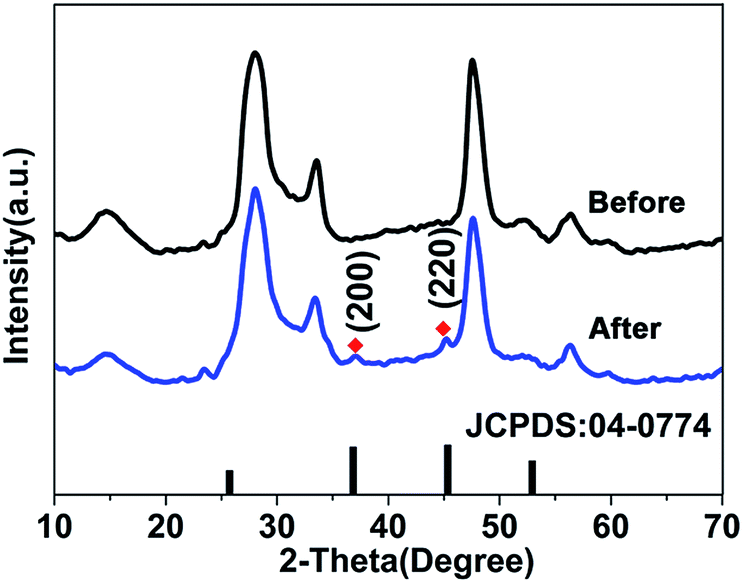 | ||
| Fig. 4 XRD patterns of ZIS MSs and ZIS MSs with the addition of AgNO3 solution (◆ indicates the peaks of the cubic Ag2S phase). | ||
3.3 Layer-by-layer XPS profiles and cation exchange mechanism
To gain more information about the inertial layers and a more conclusive mechanism of the cation exchange process in a micrometer-sized ternary crystal, we performed layer-by-layer XPS profile analysis on the samples acquired from different stages by Ar+ sputtering. Fig. 5a–c are the XPS spectra derived from the ZIS MSs after adding AgNO3 solution without heating. In accordance with the XRD patterns (Fig. 5b), the outer layers (about several nanometers) are definitely Ag2S with the atomic ratio of 2.3:1. In Fig. 5a, the two fitted peaks with lower binding energy are Ag 3d (374.7 eV for 3d3/2, 368.5 eV for 3d5/2) peaks, while the two satellites with higher binding energy are the oxidized states caused by the combination of Ag with O during synthesis. The two peaks of S 2p arise from the splitting of spin–orbital coupling.36 Fig. 5d–f are the spectra taken after sputtering for 15 min with the above sample. It demonstrates that after stripping about 15 nm of outer layers, the remainder is Zn–In–S with an atomic ratio of 1:2.04:4.2 (identical to that of ZnIn2S4), while the two weak peaks of Ag 3d still exist. Previous studies manifested that Ag+ in a cubic phase has a high diffusion coefficient similar to that in liquid (about 10−5 cm2 s−1) which is influenced significantly by temperature.28,32 So it can be inferred that, because of the high mobility, Ag+ can easily diffuse within the lattice of cubic ZnIn2S4 and doesn’t have an impact on the structure and composition of ZnIn2S4.
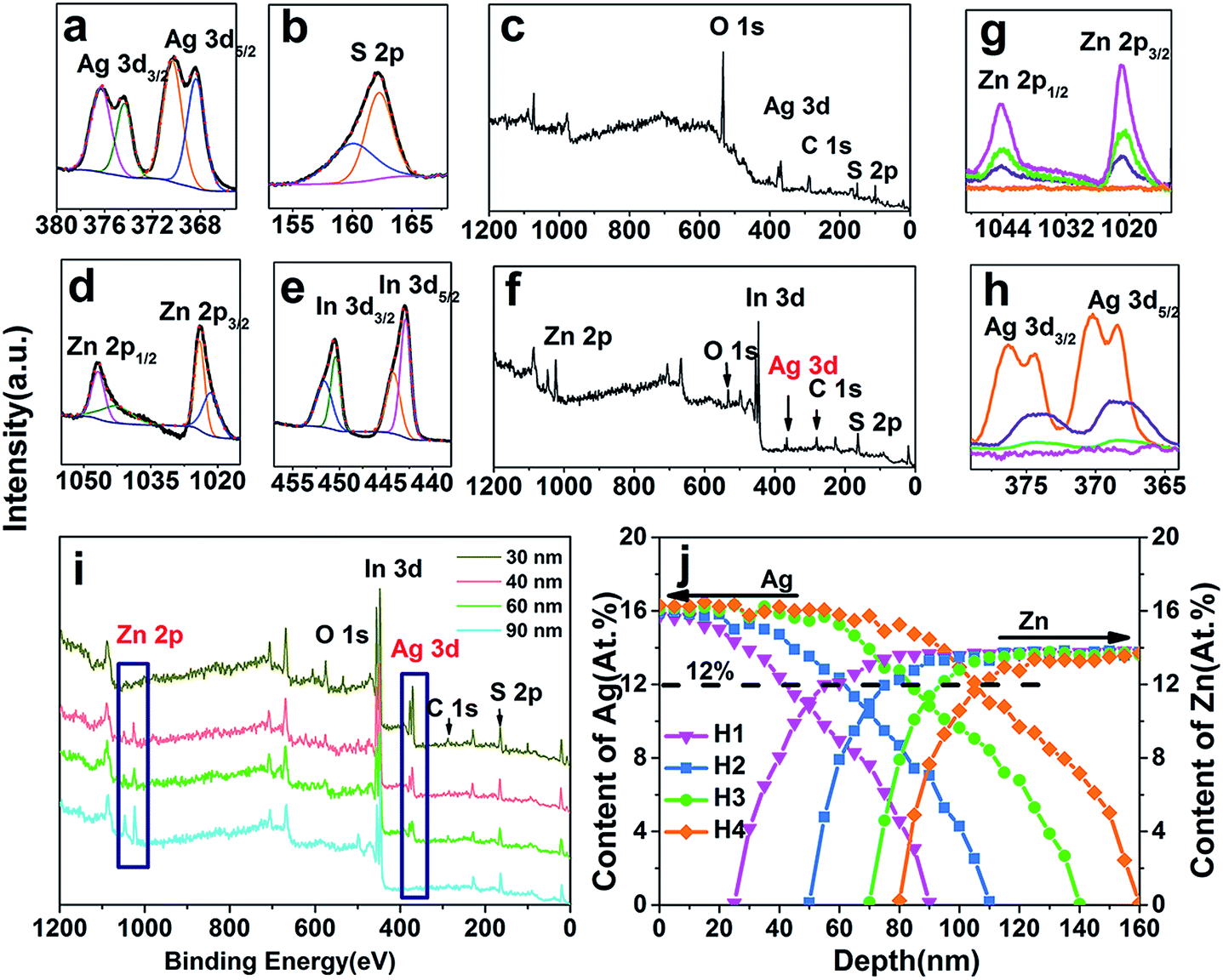 | ||
| Fig. 5 XPS profiles of (a) Ag 3d, (b) S 2p, and (c) ZIS after adding AgNO3 solution; (d) Zn 2p, (e) In 3d, and (f) ZIS MSs with sputtering of Ar+ for 15 min after adding AgNO3 solution; and high resolution spectra of (g) Zn 2p and (h) Ag 3d of (i) ZIS/AIS HMSs (sample H1) taken from different depths. (j) Content of Ag and Zn derived from different depths of samples H1–H4. The black and red curves in a, b, d and e stand for the raw data and the fitted data respectively. | ||
Further depth profile measurements were carried out to obtain more information about the components and content of the ZIS/AIS HMSs. Fig. 5i is the XPS profile taken from different depths of the representative sample H1. The spectrum for the 30 nm depth shows a typical Ag–In–S curve without Zn, while material between 40–60 nm is a gradient alloy of Ag–Zn–In–S (AZIS). The profile taken from the depth of 90 nm is the typical spectrum of ZnIn2S4. With the deepening of the sampling depth, the intensity of Ag 3d (Fig. 5h) gradually decreased along with the gradual increase of Zn 2p (Fig. 5g). Meanwhile, the oxidation satellites reasonably disappear while the position of Ag 3d3/2 and 3d5/2 stay unchanged, declaring the reduction of O in the inner microspheres.
Fig. 5j shows a plot of the content of Ag and Zn which was derived from different depths. The distribution of the Ag content is in good agreement with the diffusion of Ag+ in the cubic phase crystals.37 Driven by the concentration gradient or chemical potential, the activated Ag+ diffused in the interstice among the S2− sublattice,32 which as a consequence caused the Ag content in sample H2 and H3 to become higher than that in sample H1 and H2 in the surface segments, and gradually decreased the diffusion rate. Zn didn’t emerge in the initial 30 nm, revealing the formation of an AIS layer. With the deepening of the detection depth, Zn increases progressively to a constant of 15.1%, which can be assigned to the content in ZnIn2S4. This clearly implies that cation exchange indeed exists inside the crystals. Groeneveld22 and Shaw et al.38 hold the viewpoint that the Frenkel defects dominant the whole exchange process in that all cation exchange occurs at the surface of crystals, and subsequently both the guest and native cations diffuse as interstitials among the lattice without exchanging within crystals. They also insisted that temperature is the vital sensitive parameter during the cation exchange process and thus strongly affects both the composition and the elemental distribution profile of crystals. The most remarkable difference between organic and aqueous solutions, however, seems to be the form of guest cations, namely, cations in organic solution must bond to organic ligands to form a metal–ligand complex to stably exist, while in aqueous solution they exist as bare ions without any ligand which make them easier to incorporate into crystals and exchange with native cations. In this case, both XRD and XPS analyses clarify that cation exchange can occur in the interior of materials.
Additional information was found showing no distinct decrease in Zn until the content of Ag was more than 12% (black dashed line in Fig. 5j), i.e. the critical concentration, declaring that a sufficient concentration of Ag+ is the essential condition for the exchange reaction. In order to verify this point, another control experiment was carried out. We only changed the way of adding the Ag precursor, that is, the Ag precursor of reaction A was added at a flow of 200 μL min−1 by a peristaltic pump, while the Ag precursor of reaction B was added into solution via a single injection, and the content and concentration of AgNO3 and other conditions remained unchanged. After reaction for 3 h, both products were measured using XRF. Table 2 clearly shows that products from the continuous addition don’t show a decrease in the Zn content, while samples from a single injection obviously show a reduction in Zn from 1 to 0.84 of the composition while the content of Ag is equal in both samples. This result suggests that despite the same content of Ag precursors, cation exchange of reaction A didn’t occur because the continuously added Ag+ gradually diffused into the lattice of ZIS as uniform interstitials, just like doping, thereby the concentration of Ag+ could not reach the critical concentration, and Zn2+ could not be replaced by Ag+ as a result. On the contrary, the single injection of the Ag precursor ensured a sufficient amount of Ag+, and cation exchange could be accomplished.
| Sample | Content/wt% | Composition | |||
|---|---|---|---|---|---|
| Zn | Ag | In | S | ||
| Original ZIS | 14.74 | — | 55.06 | 30.20 | ZnIn2.13S4.19 |
| Reaction A | 13.79 | 4.87 | 52.2 | 28.71 | Zn0.98Ag0.21In2.13S4.17 |
| Reaction B | 12.07 | 5.21 | 53.46 | 29.26 | Zn0.84Ag0.22In2.12S4.16 |
So during the synthesis process, a moderate temperature of 90 °C can improve the activation energy of Ag+, and thereby allow for the incorporation and diffusion in ZIS MSs. Further heating (150 °C) can facilitate the exchange between Ag+ and Zn2+ thus favoring the reaction toward our expected product. We define the diffusion depth of Ag+ as the reaction front, and the depth from the surface to the place where Zn appears as the thickness of AIS. As shown in Table 3, the thickness of the AIS layer increased with Ag content undoubtedly.
| Sample | Thickness of AIS/nm | Thickness of ZAIS/nm | Depth of reaction front/nm |
|---|---|---|---|
| H1 | 25 | 65 | 90 |
| H2 | 50 | 60 | 110 |
| H3 | 70 | 70 | 140 |
| H4 | 80 | 80 | 160 |
On the basis of the XPS analysis and XRD studies above, a summary for the cation exchange mechanism of ZIS to AIS and AIS to Ag2S in aqueous solution is illustrated in Scheme 1. Upon adding the Ag precursor, Ag+ cations enter into the lattice of ZIS as interstitials and diffuse around Zn2+ and In3+. Within the cubic ZIS lattice, In3+ ions bind more S2− ions than Zn2+ ions. So under the same concentration of Ag+, Zn2+ is comparatively more easily electrostatically perturbed by Ag+ than In3+. From the viewpoint of electromagnetism, due to the same order of magnitude between the electrostatic energy of Ag+ (uniform charge density model: 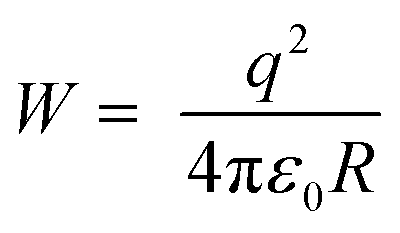 , where W is the energy, ε0, q and R is vacuum permittivity, charge and radius of the cation, respectively) and the interaction energy of the Zn–S bond (
, where W is the energy, ε0, q and R is vacuum permittivity, charge and radius of the cation, respectively) and the interaction energy of the Zn–S bond (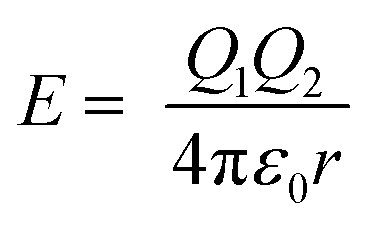 , where E is the interaction energy, Q1 and Q2 are the charges of the two particles, respectively, and r is the distance between the two particles), Ag+ has a certain probability to break the equilibration between Zn and S, and thus surrounds Zn2+. When the amount of Ag+ reaches the critical concentration, there is enough Ag+ around and Zn2+ is in the center of its regular tetrahedron cell just like a “screened” ion such that when it leaves the initial site, one of the Ag+ cations occupies the pristine site of Zn2+. On the other hand, Ag has a larger electronegativity value (1.93) than Zn (1.65) and In (1.78),31 which means that Ag is more likely to bind with S so that the energy of the overall crystal system decreases to a more stable state. As a comprehensive consequence, cation exchange between Ag+ and Zn2+ happens. Because of the concentration gradient, Zn2+ moves out of the crystals and binds to methanol finally. Thus, a cubic AIS phase is acquired. With the propagation of the reaction front and the migration of Ag+ under the condition of heating, the numbers of Ag+ around In3+ increased till they are sufficient to smash the constraint of S2− in the regular octahedron and exchange with In3+. Because of the high mobility, Ag+ in the tetrahedron rearranges to establish a structural equilibrium and Ag2S with a body-centered cubic unit cell is secured. In short, there are two stages during the cation exchange, that is (i) incorporation and diffusion of guest cations (which is analogous to doping), and (ii) bonds disrupting along with in situ exchange, which is affected by mobility and concentration, respectively. This mechanism not only elucidates the generation of Ag2S in the outer layers of ZIS without heating, but also explains the more difficult and slow reverse exchange reaction between CdS and Ag2S.23
, where E is the interaction energy, Q1 and Q2 are the charges of the two particles, respectively, and r is the distance between the two particles), Ag+ has a certain probability to break the equilibration between Zn and S, and thus surrounds Zn2+. When the amount of Ag+ reaches the critical concentration, there is enough Ag+ around and Zn2+ is in the center of its regular tetrahedron cell just like a “screened” ion such that when it leaves the initial site, one of the Ag+ cations occupies the pristine site of Zn2+. On the other hand, Ag has a larger electronegativity value (1.93) than Zn (1.65) and In (1.78),31 which means that Ag is more likely to bind with S so that the energy of the overall crystal system decreases to a more stable state. As a comprehensive consequence, cation exchange between Ag+ and Zn2+ happens. Because of the concentration gradient, Zn2+ moves out of the crystals and binds to methanol finally. Thus, a cubic AIS phase is acquired. With the propagation of the reaction front and the migration of Ag+ under the condition of heating, the numbers of Ag+ around In3+ increased till they are sufficient to smash the constraint of S2− in the regular octahedron and exchange with In3+. Because of the high mobility, Ag+ in the tetrahedron rearranges to establish a structural equilibrium and Ag2S with a body-centered cubic unit cell is secured. In short, there are two stages during the cation exchange, that is (i) incorporation and diffusion of guest cations (which is analogous to doping), and (ii) bonds disrupting along with in situ exchange, which is affected by mobility and concentration, respectively. This mechanism not only elucidates the generation of Ag2S in the outer layers of ZIS without heating, but also explains the more difficult and slow reverse exchange reaction between CdS and Ag2S.23
In the experimental process, methanol and In3+ were both introduced to facilitate the generation of AIS content and reduce the Ag2S, due to the stronger bond between methanol and divalent Zn2+ cations which favors the reaction.39 According to the proposed mechanism, we designed a new approach to control the exchange rate and progress, and by this, floriated AIS MSs and Ag2S MSs starting from ZIS MSS were obtained (ESI Fig. S4†). These results demonstrate a fundamental feature of cation exchange in ternary crystals, that both the mobility and the concentration of cations can remarkably tailor the composition and distribution of the product. The work provides a model description of the transformation between different ternary crystals and the conversion from ternary to binary crystals, suggesting that cation exchange is a versatile protocol which can be applied at both the nano- and micro-scale.
3.4 UV-vis-NIR absorption and band structure
Diffuse reflectance spectra were used for characterizing the optical properties of materials. As shown in Fig. 6, a steep absorption edge for the ZIS MSs at around 525 nm is observed, manifesting an intrinsic state-related transition from valence band (VB) to conduction band (CB). The curves of the ZIS/AIS MSs similarly show the absorption of excitons with red shifts of the absorption onset from 593 to 810 nm, corresponding to the band gap of 2.09 to 1.53 eV (Table 4). This demonstrates the narrowing of the band gap because of the incorporation of Ag into the valence band of AIS.40–42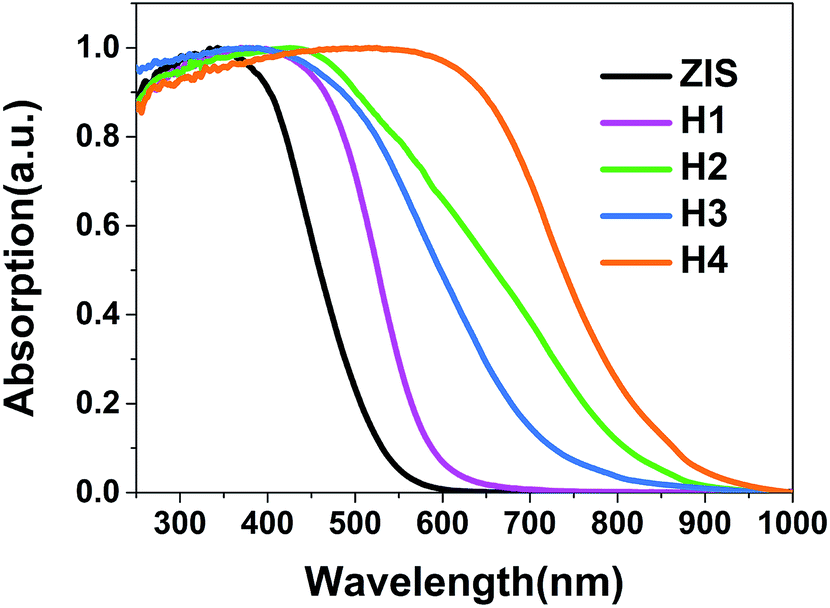 | ||
| Fig. 6 UV-vis-NIR diffuse reflectance absorption spectra of each sample. | ||
| Sample | Absorption edge (nm) | Band gap (eV) | BET specific surface area (m2 g−1) | Pore size (nm) |
|---|---|---|---|---|
| ZIS | 525 | 2.36 | 109.16 | 3.1 |
| H1 | 593 | 2.09 | 103.83 | 3.0 |
| H2 | 685 | 1.81 | 99.74 | 3.2 |
| H3 | 763 | 1.62 | 95.23 | 3.1 |
| H4 | 810 | 1.53 | 92.06 | 3.0 |
In order to elucidate the charge separation process of the as-prepared ZIS/AIS HMSs, the positions of the bands for ZIS can be calculated using Mulliken electronegativity and band gap values by the following equations:43
| EVB = χ − Ee + 0.5Eg | (1) |
| ECB = EVB − Eg | (2) |
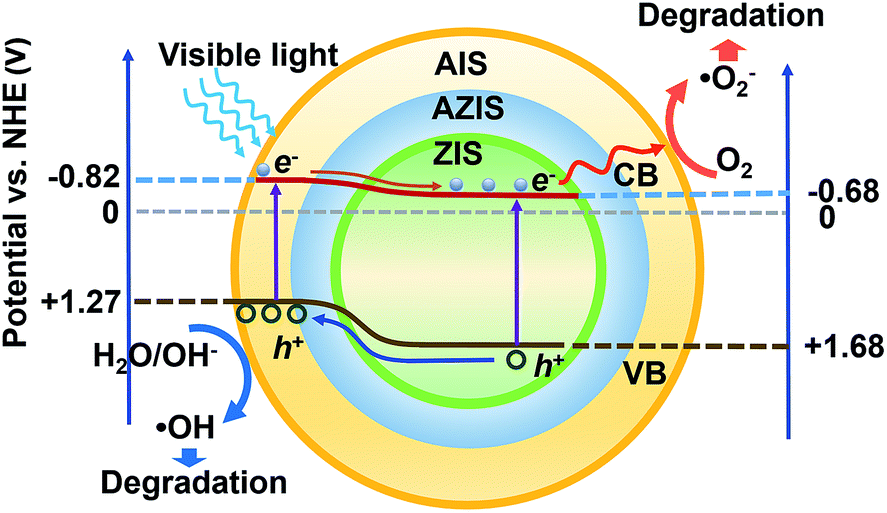 | ||
| Scheme 2 Proposed photocatalytic mechanism for the visible light-driven photocatalytic dye degradation. | ||
Under visible light illumination, electrons are excited to the CB of AIS, and inject into the CB of ZIS since the ECB is lower than that of AIS. Simultaneously, the photogenerated holes are injected into the VB of AIS, and directly oxidize OH− or H2O molecules because the EVB is more positive than E0(O2/H2O) (+1.23 V vs. NHE), such that ˙OH active species are formed. In the meantime, the photogenerated electrons can migrate to the CB of ZIS and reduce O2 to produce a superoxygen radical (˙O2−) due to its more negative potential than ˙O2−/O2 (−0.33 V vs. NHE). Subsequently, some of ˙O2− continues to react with H+ to produce H2O2, which can also degrade dye. Because of their wide absorption in the visible light region compared to ZIS and the perfect separation of photogenerated carriers, the ZIS/AIS HMSs can provide a pronounced degradation efficiency. From the perspective of energy level matching and electrochemistry, this kind of band structure can also be used as a light absorber to fabricate solar cells or for hydrogen evolution.
3.5 BET analysis and photocatalytic dye degradation
It is of great importance to evaluate the surface properties of our prepared ZIS/AIS HMSs before applying them to the degradation of a dye. Nitrogen adsorption–desorption results shown in Fig. 7a demonstrate a type IV isotherm with a H3 hysteresis loop in a relatively lower pressure region, suggesting the mesoporous nature of the samples.11 The broad hysteresis loop illustrates a wide pore size distribution of the ZIS MSs and ZIS/AIS HMSs. Fig. 7b shows the pore size distribution graphs of the samples with an average size around 3 nm (Table 4), consistent with the SEM characterization. | ||
| Fig. 7 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution plots of ZIS and ZIS/AIS samples; temporal UV-vis absorption spectra of pre-adsorption (PA) and during the photocatalytic degradation of Rhodamine B in aqueous (c) ZIS MS and (d) ZIS/AIS HMS (sample H2) suspensions (insets: photographs of Rhodamine B solution taken at 10 to 50 min during irradiation); (e) visible light photocatalytic activities for Rhodamine B degradation and (f) degradation rate after irradiation for ZIS and ZIS/AIS samples. | ||
We chose Rhodamine B (RhB) as a representative cationic dye for the photodegradation test. During the establishment of an adsorption/desorption equilibrium, RhB molecules were bound to the surface of the ZIS MSs and ZIS/AIS HMSs via –N(Et)2 groups, because positively charged RhB molecules have a strong interaction with the ZIS MSs and ZIS/AIS HMSs which have many negative charged S sites.11 In the process of photodegradation, however, the ZIS/AIS HMSs exhibited a higher degradation rate (up to 99.2% for sample H2) than that of the ZIS MSs (52.2%) as shown in Fig. 7f, although they possess a smaller surface area than that of the ZIS MSs (Table 4). The slight blue shifts of the absorption peaks may be the N-de-ethylated intermediates formed in a stepwise manner during the photodegradation.12 Because of the thinner layers of AIS compared to the wavelength of visible light, visible light of various wavelengths can be well absorbed by Ag–In–S, Ag–Zn–In–S and Zn–In–S sections, and a marvelous degradation rate is achieved. Further XRD measurements of the ZIS/AIS HMSs before and after 10 photodegradation cycles show an essentially similar pattern, manifesting the considerable photostability of the HMSs (shown in ESI Fig. S5†).
Intriguingly, the degradation rate of sample H3 and H4 is lower than that of H1 and H2, even below the ZIS MSs in the case of H4. This may be the result of the continuous incorporation of Ag+ ions. Since the VB of AIS is composed of S 3p and Ag 4d,40,42,45–47 the continuously increased Ag content (Fig. 5j) may lift the VB potential until E0(O2/H2O), thus ˙OH active species cannot be formed. What’s more, with the progression of the reaction and thickening of the AIS layer, the length of the migration path for both photogenerated carriers grew correspondingly, increasing the recombination probability and therefore reducing the number of active species. So, in the light of the results of photodegradation and XPS analysis, a conclusion could be made that an appropriate thickness (about 50 nm) of AIS can obviously enhance the rate of degradation, where too thin or too thick compositions may result in the reduction of photodegradation efficiency. This structure may also be used as a light absorber to fabricate solar cells.
4. Conclusion
In conclusion, AIS/ZIS HMSs with different compositions and band gaps were synthesized by cation exchange. Based on the SEM, TEM, XRD and XPS analyses, a possible mechanism for cation exchange within ternary crystals and between ternary to binary crystals was proposed. According to the UV-vis-NIR absorption spectra, an enhanced photodegradation mechanism was discussed, revealing that an appropriate thickness in the AIS layer can be favorable for the generation of active species. Furthermore, a photodegradation test with a dye was performed, and the ZIS/AIS HMSs demonstrated a marvelous degradation rate up to 99% under the irradiation of visible light. Our work is not only useful for the synthesis of heteromaterials between different band structures but also provides some insight into the design for tailoring a multi-composite to deliver a high photocatalytic performance, which may provide new opportunities for the evolution of hydrogen and photovoltaic applications.Acknowledgements
We are sincerely grateful to Mr Jufeng Huang, Dr Weijia Shao, Prof. Ming Gong, and Dr Jie Tian for the helpful discussions and technical assistance. We also appreciate Mr Asad Ali for his useful suggestion during the revision of the paper. This study is financially supported by the National Natural Science Foundation of China (No. 51272246) and Scientific and Technological Research Foundation of Anhui (No. 12010202035).References
- Z. Feng, P. Dai, X. Ma, J. Zhan and Z. Lin, Appl. Phys. Lett., 2010, 96, 013104 CrossRef.
- Y. Sheng, X. Tang and J. Xue, J. Mater. Chem., 2012, 22, 1290–1296 RSC.
- J. Song, T. Jiang, T. Guo, L. Liu, H. Wang, T. Xia, W. Zhang, X. Ye, M. Yang, L. Zhu, R. Xia and X. Xu, Inorg. Chem., 2015, 54, 1627–1633 CrossRef CAS PubMed.
- W. Xiang, C. Xie, J. Wang, J. Zhong, X. Liang, H. Yang, L. Luo and Z. Chen, J. Alloys Compd., 2014, 588, 114–121 CrossRef CAS.
- T. Torimoto, T. Adachi, K.-i. Okazaki, M. Sakuraoka, T. Shibayama, B. Ohtani, A. Kudo and S. Kuwabata, J. Am. Chem. Soc., 2007, 129, 12388–12389 CrossRef CAS PubMed.
- B. Mao, C.-H. Chuang, F. Lu, L. Sang, J. Zhu and C. Burda, J. Phys. Chem. C, 2013, 117, 648–656 CAS.
- K. J. Hong, J. W. Jeong, T. S. Jeong, C. J. Youn, W. S. Lee, J. S. Park and D. C. Shin, J. Phys. Chem. Solids, 2003, 64, 1119–1124 CrossRef CAS.
- S. P. Hong, H. K. Park, J. H. Oh, H. Yang and Y. R. Do, J. Mater. Chem., 2012, 22, 18939–18949 RSC.
- S. Shionoya and Y. Tamoto, J. Phys. Soc. Jpn., 1964, 19, 1142–1149 CrossRef CAS.
- B. Chai, T. Peng, P. Zeng, X. Zhang and X. Liu, J. Phys. Chem. C, 2011, 115, 6149–6155 CAS.
- Y. Chen, R. Huang, D. Chen, Y. Wang, W. Liu, X. Li and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2273–2279 CAS.
- Z. Chen, D. Li, W. Zhang, Y. Shao, T. Chen, M. Sun and X. Fu, J. Phys. Chem. C, 2009, 113, 4433–4440 CAS.
- F. Fang, L. Chen, Y.-B. Chen and L.-M. Wu, J. Phys. Chem. C, 2010, 114, 2393–2397 CAS.
- X. Gou, F. Cheng, Y. Shi, L. Zhang, S. Peng, J. Chen and P. Shen, J. Am. Chem. Soc., 2006, 128, 7222–7229 CrossRef CAS PubMed.
- T. Jiang, J. Song, H. Wang, X. Ye, H. Wang, W. Zhang, M. Yang, R. Xia, L. Zhu and X. Xu, J. Mater. Chem. B, 2015, 3, 2402–2410 RSC.
- Y. Yu, G. Chen, G. Wang and Z. Lv, Int. J. Hydrogen Energy, 2013, 38, 1278–1285 CrossRef CAS.
- X. Yuan, R. Ma, W. Zhang, J. Hua, X. Meng, X. Zhong, J. Zhang, J. Zhao and H. Li, ACS Appl. Mater. Interfaces, 2015, 7, 8659–8666 CAS.
- W. Zhang, Q. Lou, W. Ji, J. Zhao and X. Zhong, Chem. Mater., 2014, 26, 1204–1212 CrossRef CAS.
- K.-W. Cheng and S.-C. Wang, Mater. Chem. Phys., 2009, 115, 14–20 CrossRef CAS.
- Q. A. Akkerman, A. Genovese, C. George, M. Prato, I. Moreels, A. Casu, S. Marras, A. Curcio, A. Scarpellini and T. Pellegrino, ACS Nano, 2015, 9, 521–531 CrossRef CAS PubMed.
- B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759–19770 CAS.
- E. Groeneveld, L. Witteman, M. Lefferts, X. Ke, S. Bals, G. van Tendeloo and C. de Mello Donega, ACS Nano, 2013, 7, 7913–7930 CrossRef CAS PubMed.
- R. Gui, J. Sun, D. Liu, Y. Wang and H. Jin, Dalton Trans., 2014, 43, 16690–16697 RSC.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef CAS PubMed.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- W. van der Stam, A. C. Berends, F. T. Rabouw, T. Willhammar, X. Ke, J. D. Meeldijk, S. Bals and C. de Mello Donega, Chem. Mater., 2015, 27, 621–628 CrossRef CAS.
- K. Miszta, D. Dorfs, A. Genovese, M. R. Kim and L. Manna, ACS Nano, 2011, 5, 7176–7183 CrossRef CAS PubMed.
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- J. Park and S.-W. Kim, J. Mater. Chem., 2011, 21, 3745–3750 RSC.
- M. D. Regulacio, K. Y. Win, S. L. Lo, S.-Y. Zhang, X. Zhang, S. Wang, M.-Y. Han and Y. Zheng, Nanoscale, 2013, 5, 2322–2327 RSC.
- J. Dean, Mater. Manuf. Processes, 1990, 5, 687–688 CrossRef.
- M. A. Hamilton, A. C. Barnes, W. S. Howells and H. E. Fischer, J. Phys.: Condens. Matter, 2001, 13, 2425 CrossRef CAS.
- W. Jost, J. Chem. Phys., 1933, 1, 466–475 CrossRef CAS.
- K. Li, B. Chai, T. Peng, J. Mao and L. Zan, ACS Catal., 2013, 3, 170–177 CrossRef CAS.
- T. Ogawa, T. Kuzuya, Y. Hamanaka and K. Sumiyama, J. Mater. Chem., 2010, 20, 2226–2231 RSC.
- Y. Du, B. Xu, T. Fu, M. Cai, F. Li, Y. Zhang and Q. Wang, J. Am. Chem. Soc., 2010, 132, 1470–1471 CrossRef CAS PubMed.
- H. Okazaki, J. Phys. Soc. Jpn., 1967, 23, 355–360 CrossRef CAS.
- D. Shaw, J. Cryst. Growth, 1988, 86, 778–796 CrossRef CAS.
- R. Klenk, J. Klaer, R. Scheer, M. C. Lux-Steiner, I. Luck, N. Meyer and U. Rühle, Thin Solid Films, 2005, 480–481, 509–514 CrossRef CAS.
- M. Dai, S. Ogawa, T. Kameyama, K.-i. Okazaki, A. Kudo, S. Kuwabata, Y. Tsuboi and T. Torimoto, J. Mater. Chem., 2012, 22, 12851–12858 RSC.
- M.-l. Dai, K.-i. Okazaki, A. Kudo, S. Kuwabata and T. Torimoto, Electrochemistry, 2011, 790–792 CrossRef CAS.
- W. Xiang, C. Xie, J. Wang, J. Zhong, X. Liang, H. Yang, L. Luo and Z. Chen, J. Alloys Compd., 2014, 588, 114–121 CrossRef CAS.
- B. Luo, D. Xu, D. Li, G. Wu, M. Wu, W. D. Shi and M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 17061–17069 CAS.
- Y. Xu and M. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406–13413 CrossRef CAS PubMed.
- I. Tsuji, H. Kato and A. Kudo, Angew. Chem., 2005, 117, 3631–3634 CrossRef.
- I. Tsuji, H. Kato and A. Kudo, Chem. Mater., 2006, 18, 1969–1975 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of broken ZIS MSs and AIS catalyst. STEM-EDS line scan of sample H1, SEM, HRTEM, XRD and method of prepared AIS and Ag2S MSs. See DOI: 10.1039/c5ra17591g |
| This journal is © The Royal Society of Chemistry 2015 |