
Stimuli responsive reversible high contrast off–on fluorescence switching of simple aryl-ether amine based aggregation-induced enhanced emission materials†
Abstract
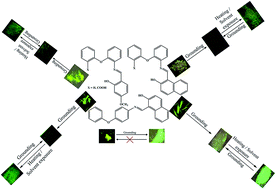
Aryl-ether amine based simple Schiff base molecules (1–5) showed aggregation induced enhanced emission (AIEE) in the solid state and rare stimuli responsive fluorescence off–on switching. The hard grounding of 1 resulted in irreversible fluorescence tuning from greenish-yellow to green. Heating or solvent exposure did not result in any fluorescence reversibility. Interestingly, the hard grounding of 2–5 led to the quenching of the solid state fluorescence and heating/solvent exposure produced clear bright fluorescence. Importantly, 2–5 exhibited reversible off–on fluorescence switching for several cycles without significant change in fluorescence intensity. It is noted that the turn-on fluorescence of 2–5 was slightly blue shifted compared to the initial solids. PXRD studies suggest that the switching of dark to bright fluorescence and vice versa of 2–5 is due to the reversible change of crystalline to amorphous phase and more planarization of the twisted structure. Single crystal analysis of 1 and 5 confirmed the twisted molecular conformation and strong intermolecular interactions in the crystal lattice, which led to AIEE by restricting free rotation and rigidifying the fluorophores in the solid state. The high contrast dark and bright fluorescence switching of 2–5 could be of potential interest in optoelectronic applications.
 Please wait while we load your content...
Please wait while we load your content...

