
Antioxidant and hepatoprotective effects of a pigment–protein complex from Chlorella vulgaris on carbon tetrachloride-induced liver damage in vivo
Abstract
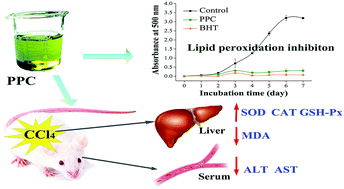
The antioxidant and hepatoprotective activities of pigments in the natural form of a pigment–protein complex were investigated. Pigment–protein complex (PPC) was isolated from Chlorella vulgaris through thylakoid protein solubilization, anion exchange chromatography and gel filtration chromatography. PPC possessed a chlorophyll : lutein : protein ratio of 94 : 153 : 100 (w/w/w) according to HPLC and spectrophotometric analysis. Various antioxidant evaluation systems were used to evaluate the antioxidant activity of PPC in vitro. Results showed that PPC exhibited significant DPPH radical scavenging activity with an IC50 of 313 μg mL−1. Distinct Fe2+ ion chelating activity, reducing capacity and lipid peroxidation inhibition activity were observed at a concentration of 1 mg mL−1. For the hepatoprotective effects, administration of PPC at different concentrations (50 and 100 mg kg−1 BW) could significantly decrease the carbon tetrachloride-induced elevation of the hepatosomatic index. Increased serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were attenuated with PPC pretreatment. In addition, PPC effectively restored suppressed hepatic superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) activities. Moreover, PPC significantly reduced the formation of malondialdehyde (MDA). The results obtained from this study clearly verified the hepatoprotective effect of PPC in CCl4-induced hepatotoxicity in vivo, suggesting that PPC has the potential to be exploited as a dietary supplement against free radical oxidation which could enhance resistivity against oxidative stress in the human body.
 Please wait while we load your content...
Please wait while we load your content...

