
Development of an efficient magnetically separable nanocatalyst: theoretical approach on the role of the ligand backbone on epoxidation capability†
Abstract
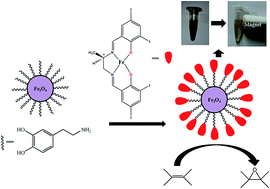
Three chiral Schiff base ligands H2L1, H2L2, H2L3 have been synthesized by treating (R)-1,2-diaminopropane separately with 3,5-dichlorosalicylaldehyde, 3,5-dibromosalicylaldehyde and 3,5-diiodosalicylaldehyde, respectively. Three new asymmetric FeIII complexes, namely, FeL1Cl (1), FeL2Cl (2), FeL3Cl (3) have been prepared from their corresponding ligands. The crystal structure of 2 reveals that the complexes are mononuclear in nature. Circular dichroism (CD) studies suggest that the ligands and their corresponding complexes contain an asymmetric center. The catalytic activity of these complexes toward the epoxidation of alkenes has been investigated in the presence of iodosylbenzene (PhIO), in two solvents CH3CN and CH2Cl2. The epoxide yield suggests that the order of their catalytic efficiency is 3 > 2 > 1. This trend as well as the role of substitution on the ligand backbone on alkene epoxidation has also been confirmed by density functional theory (DFT) calculations. For further adaptation, we attached our most efficient homogeneous catalyst, 3, with surface modified magnetic nanoparticles (Fe3O4@dopa) and thereby obtained the new magnetically separable nanocatalyst Fe3O4@dopa@FeL3Cl. This catalyst has been characterized and its olefin epoxidation ability investigated in similar conditions to those used for homogeneous catalysts. The enantiomeric excess of the epoxide yield reveals the retention of chirality of the active site of Fe3O4@dopa@FeL3Cl. The catalyst can be easily recovered by magnetic separation and recycled several times without significant loss of its catalytic activity.
 Please wait while we load your content...
Please wait while we load your content...

