
Why do the structural properties of complexes formed by glucans and carbon nanotubes differ so much?†
Abstract
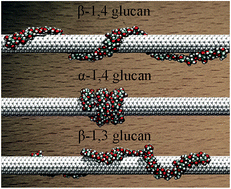
In supramolecular wrapping chemistry, polysaccharides are widely used as wrapping agents for the dissolution, dispersion and functionalization of carbon nanotubes. It is, therefore, of paramount importance to understand the effect of the topology – specifically the linkage – of the polymer chain on its spatial arrangement around the hollow tubular structure, and, hence, on the configuration and the nature of the supramolecular complex. To this end, the β-1,4, α-1,4 and β-1,3-glucans were chosen to wrap a single-walled carbon nanotube (SWCNT) as three prototypical assemblies. Molecular simulations reveal that α-1,4-glucan has the ability to wrap SWCNTs very tightly, whereas β-1,3-glucans can only form irregular helices. The calculated binding affinity of the polysaccharide to the tubular surface follows the order α-1,4 > β-1,4 > β-1,3-glucan. The differences between the three hybrids can be generally described in terms of the inherent propensity of glucans to fold into helices, the hydrophobic interaction of the polysaccharide with the SWCNT, and the formation of intramolecular hydrogen bonds within the polymer chain. The wrapping mode of the glucan chain is mainly determined by its inherent helicity. The hydrophobic interaction is the driving force for helical wrapping. Moreover, the intramolecular hydrogen-bonding interaction can stabilize ideal, compact helical scaffolds. These factors determine the conformation and the binding affinity of the polysaccharide to the SWCNT. The present results can be generalized to other polymers like DNA, and shed new light on the universal principles that underlie the formation of supramolecular complexes using wrapping agents.
 Please wait while we load your content...
Please wait while we load your content...

