
Flux growth of patterned LiCoO2 crystal arrays directly on a Pt substrate in molten LiNO3†
T. Yodaab,
N. Zettsuac,
H. Onoderaa,
Y. Mizunoa,
H. Kondob and
K. Teshima*ac
aDepartment of Environmental Science & Technology, Shinshu University, 4-17-1 Wakasato, Nagano 380-8553, Japan. E-mail: teshima@shinshu-u.ac.jp
bShinko Electric Industries Co. Ltd, 80 Oshimada-machi, Nagano 381-2287, Japan
cCenter for Energy and Environmental Science, Shinshu University, 4-17-1 Wakasato, Nagano 380-8553, Japan
First published on 2nd November 2015
Abstract
We demonstrate a new way to prepare hollow-structured LiCoO2 crystals directly on a Pt substrate for the first time through a combination of semi-additive electrodeposition of a Co core and subsequent flux growth in molten LiNO3. The reaction process was characterized by time-dependent X-ray diffraction and scanning electron microscopy. The vertically oriented crystals having a platelet shape grew densely on the Co dot surface. The crystal growth was driven by supersaturation in the same manner as the flux growth. Significantly slower oxidation of the Co core and rapid lithiation of Co3O4 lead to pore formation, which suggests that slow oxygen diffusion in the Co core is rate limiting. Galvanostatic tests revealed that the LiCoO2 crystal array exhibited typical capacity–voltage profiles with no heavy capacity loss during the first three cycles without any additives.
Introduction
Flux growth is one of the most versatile ways to obtain idiomorphic crystals at low temperatures without any thermal strain or defects from high-temperature solutions.1–3 Thus, it has many advantages over other techniques of functional material synthesis. For instance, flux growth methods provide well-faceted single crystals, which improve lithium ion secondary battery (LIB) performances. The flux of LiCl, NaCl, KCl, NaCl–KCl, and LiCl–KCl enables the growth of layered LiCoO2,4,5 Li(Ni0.8Mn0.1Co0.1)O2,6 spinel LiMn2O4,7,8 Li4Ti5O12,9,10 and olivine LiFePO4 (ref. 11–13) crystals, which show significantly enhanced electrochemical performances with high capacity, excellent cycle stability, and good rate capability. In addition, such nicely faceted crystals exhibit face-selective properties such as face-selective tolerance towards the loss of electrolyte-soluble transition metal ions in the crystals.14Flux growth techniques provide several aspects of desired crystal growth by adjustment or optimization of growth conditions. For instance, the flux selected and the additives or mineralizers used, such as OH−, F−, Cl−, affect crystal nucleation and growth, so they affect the crystal size, shape, and habit through the dissolution of the reagents by complex formation in solution. This increases the metastable region where supersaturation can occur, and alternating the viscosity of the hot solutions.1,2
Very recently, we extended the flux growth concept to a coating technique for fabricating a polycrystal film deposited directly on substrates.10,11,15 High-quality crystals were grown using a paste composed of the solutes and flux for the growth, which were coated onto the substrate prior to heating. The substrate surface was fully covered with well-defined faceted single crystals. Cross-sectional transmission electron microscopy (TEM) observation along with EDS and SAED analysis revealed that at the interface, strong bonding exits on the atomic level between the crystals and the substrate with no impurity phase. Time-dependent X-ray diffraction (XRD) and scanning electron microscopy (SEM) suggested that the crystal growth mechanism was analogous to flux growth driven by supersaturation. The initial heterogeneous nucleation and subsequent growth in a homogeneous hot solution resulted in the formation of the crystal layer on the substrate. We called this new coating methodology flux coating. The LiCoO2, Li4Ti5O12 polycrystal films showed a large capacity close to the theoretical values with no assistance from any additives such as carbon black and polyvinylidene fluoride (PVDF) under moderate C rate conditions.10,15 This indicated that Li+ ions and electrons were efficiently transferred in the electrode and interfaces during the intercalation and deintercalation reaction. Thus, we believe such additive-free electrodes potentially enhance the specific energy density per electrode volume.
For expanding the application of additive-free electrodes produced by flux coating, we propose a new way to prepare desirable additive-free electrodes directly on a substrate using a combination of electrodeposition of transition metals and flux coating. This is advantageous because electrodeposition is a powerful means of mass production of thin metal films at a low cost. Furthermore, micrometer-scale architectures can be formed via combined photolithography and a lift-off technique with electrodeposition. This process is called semi-additive electrodeposition. Macroscopic architectures of electrodes in LIBs increase the reactive surface area as well as accelerate the Li-ion transfer at the electrolyte interface, leading to high energy transfer.16–18 For instance, Pikul et al.18 demonstrated continuous microporous electrodes using a combination of colloidal self-assembly of polystyrene nanospheres as the template and electrodeposition of active materials; the resulting electrodes exhibited a micro-architecture and achieved high rate capabilities. However, to the best of our knowledge, there are no previous reports on electrodeposited Co with the fine patterns obtained by using semi-additive electrodeposition.
In this study, we demonstrate the fabrication of spatially designed LiCoO2 crystal assemblies and control their periodicity through a combination of semi-additive electroplating of metallic Co and flux growth of LiCoO2 crystals in molten LiNO3, which realizes uniform, low-cost, and large-scale production of designed LIBs electrodes. We characterized the reaction process using time-dependent XRD and SEM. We also characterized their primitive electrochemical properties for use as cathodes for LIBs using galvanostatic tests.
Experimental section
A mixed aqueous solution containing 350 g L−1 of cobalt sulfate (CoSO4·H2O 350 g L−1), 30 g L−1 of cobalt chloride (CoCl2·6H2O), 30 g L−1 of boric acid, and malonic acid as an organic acid having carboxyl groups was used for Co plating. The pH and temperature of the mixed solution were maintained at 4.0 and 50 °C, respectively, during the plating. Ammonia water and an aqueous solution of sulfuric acid were used as pH adjusters. A photolithographic technique followed by a lift-off process was applied to prepare the templates for the fabrication of the spatially designed Co microstructures. A dry film developed by Hitachi Chemical Corporation was used as a photoresist. The photoresist film was used to laminate a clean Pt substrate. The thickness of the photoresist was ∼15 μm. After irradiation with UV light, the unreacted resist film was developed using an aqueous solution of sodium carbonate. The plating process was progressed in two steps by tuning the current density first to 6.0 A dm−2 for 1 min and then to 2.0 A dm−2. The thicknesses of the Co layers were controlled to be 7 μm. Finally, the dry film was removed carefully by using n-methyl pyrolidone. Spatially patterned electrodeposited Co cores were converted to LiCoO2 crystals in molten LiNO3 at 500 °C. Platinum substrates with 0.1 mm thickness were used in this study as a priority in validating the evidence of our demonstration. Lithium nitrate was used as both a Li source and a flux for the growth. The concentration of the aqueous LiNO3 solution was 30 wt%. Ten milliliters of the aqueous LiNO3 solution was spread onto the Co layer with dimensions of 10 × 10 mm by drop casting. The corresponding solute concentration was 2.65 wt%. The substrate was heated to 500 °C at a rate of 1000 °C h−1 in an electric furnace. After maintain the temperature for 10 h, the crucible was cooled to 100 °C at rate of 200 °C h−1. The heated substrates were then naturally cooled to room temperature in the furnace. Finally, the substrates were immersed in warm water at 80 °C for 30 min to remove the remaining flux. The washed substrate was dried at 100 °C under air. The morphology and average size of the grown LiCoO2 crystals were characterized using field-emission scanning electron microscopy (FE-SEM, JEOL, JSM-7600F). The acceleration voltage was of 15 kV. The samples for the cross-sectional SEM observation were prepared by focused ion beam milling (FIB, JEOL, JIB-4601F) with a Ga-ion source. The FIB milling was performed at 30 kV. A tungsten layer was deposited on the sample surfaces as a protection layer prior to milling. The phases and structures of the crystals were identified using XRD analysis with Cu Kα radiation. The X-ray diffractometer (RIGAKU, MiniflexII) was operated at 30 kV and 20 mA, with 2θ = 10–80°. The charge–discharge characteristics of the LiCoO2 crystal layer as an additive-free cathode were studied using a coin-type cell (R2032 type). A galvanostatic charge–discharge test was conducted using a battery tester (HOKUTO DENKO, HJ1001SD8) with a voltage window of 2.5–4.2 at setting current rate. The LiCoO2 crystals on the Pt substrates were annealed at 700 °C for 1 h under air to promote crystallization prior to cell assembly. A mixed solution of ethylene carbonate and dimethyl carbonate containing 1 M of LiPF6 was used as an electrolyte. Li metal foil was used as an anode. A polypropylene separator (Celgard separator #2400) was used for the suppression of direct contact of each electrode. The coin-type LIBs were assembled in an Ar-filled glove box (MIWA MFG Co. Ltd.) with a controlled atmosphere containing less than 1 ppm of H2O and O2.Results and discussion
In general, Co can be electrodeposited from the Co2+-containing solutions stabilized with sulfate chloride, ammonium sulfamate, and fluoborate.19–23 As a result, Co plating films hardly adhere to electrodes owing to the high interface stress. Therefore, the Ni plating process was commonly employed for strain release before Co deposition was initiated. The thin soft Ni layer worked as a buffer. From the viewpoint of material synthesis, however, the external metal species becomes a potential source of impurity or sub-phase formation. Thus, we demonstrated two-step Co electrodeposition under different current densities, without employing the Ni layer.Typical SEM images of the Co microstructures are shown in Fig. 1(a and b). Ultra-fine Co dot matrices arranged in a 4-fold symmetry were uniformly formed with 10 × 10 cm dimensions. Fig. 2(a and e) show the changes in XRD profiles of the microstructured Co electrodeposits after heating in molten LiNO3 flux at 500 °C for 50 h. All diffraction lines in Fig. 2(e) can be assigned as per the International Centre for Diffraction Data (ICDD) for LiCoO2 (PDF 75-0532), which belongs to an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and the Pt substrate. The lattice parameters were determined to be a = 2.819 and c = 14.101 Å. These values closely agree with previous data.4,24–27 LiNO3 flux decreased the reaction temperature drastically to 500 °C, lower than that of solid-state-reaction processes. Notably, the relative intensities of the diffraction lines at 2θ = 18.93/45.26, assigned to 003LiCoO2/104LiCoO2, were less than that as per the ICCD data, suggesting that the LiCoO2 crystals were dominantly covered with 104LiCoO2 planes. As shown in Fig. 1(c and d), SEM observation showed the Co dots were uniformly transformed into hemispherical dots that maintained their original periodicity. The numerous small crystals with platelet shapes were formed densely in the hemispherical dots. It is appeared that all individual crystals were oriented perpendicularly toward the surface. The platelet shape of the LiCoO2 crystal is known to be the result of a thermodynamically favored state produced under an oxidative atmosphere.4,5,24–27 The wide flat plate of the crystal can be assigned to the c-plane, and their sharp and asymmetrical side faces are surrounded by a, b faces.
m space group and the Pt substrate. The lattice parameters were determined to be a = 2.819 and c = 14.101 Å. These values closely agree with previous data.4,24–27 LiNO3 flux decreased the reaction temperature drastically to 500 °C, lower than that of solid-state-reaction processes. Notably, the relative intensities of the diffraction lines at 2θ = 18.93/45.26, assigned to 003LiCoO2/104LiCoO2, were less than that as per the ICCD data, suggesting that the LiCoO2 crystals were dominantly covered with 104LiCoO2 planes. As shown in Fig. 1(c and d), SEM observation showed the Co dots were uniformly transformed into hemispherical dots that maintained their original periodicity. The numerous small crystals with platelet shapes were formed densely in the hemispherical dots. It is appeared that all individual crystals were oriented perpendicularly toward the surface. The platelet shape of the LiCoO2 crystal is known to be the result of a thermodynamically favored state produced under an oxidative atmosphere.4,5,24–27 The wide flat plate of the crystal can be assigned to the c-plane, and their sharp and asymmetrical side faces are surrounded by a, b faces.
 | ||
| Fig. 1 Changes in the 45°-tilted FE-SEM images of the Co dot arrays directly deposited on the Pt substrate: (a and b) as prepared Co dot with average diameter of 7 μm (c and d) after heating at 500 °C for 50 h in a molten LiNO3. | ||
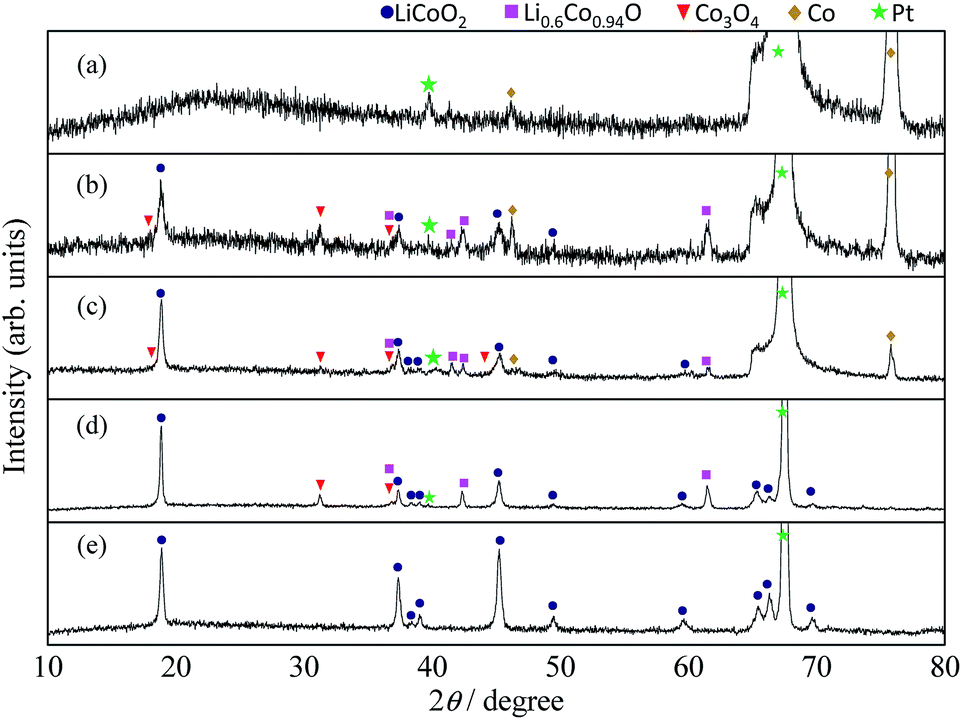 | ||
| Fig. 2 Changes in the XRD profiles of the Co dot directly deposited on Pt substrate: (a) as prepared (b) 500 °C, (c) 500 °C for 1 h (d) 500 °C for 10 h, (e) 500 °C for 50 h. | ||
To gain insight into the reaction mechanism responsible for LiCoO2 phase formation, time-dependent ex situ XRD measurements and SEM observation were carried out. Fig. 2(a–e) show the changes in the XRD profiles of the Co dot matrix having 7 μm diameter as a function of reaction time at 500 °C. The LiCoO2 phase was formed as the primary phase after 1 h despite the coexistence of metallic Co, Li0.6Co0.94O and Co3O4. The diffraction lines attributed to Co metal completely disappeared after 10 h. Instead, both the diffraction intensity of Li0.6Co0.94O and Co3O4 increased. Further increase in the reaction time to 50 h resulted in the formation of LiCoO2 as a single component. The formation of the CoO phase as an intermediate5 was not detected by ex situ XRD measurement during the whole reaction process. This suggests that LiNO3 oxidizes Co to be Co3O4 with an assistance of Co as catalysts for the decomposition of LiNO3, and Li+ and NO2 were released in hot melt at 500 °C, as following plausible reaction scheme:
| 4LiNO3 + Co → 4NO2 + Co3O4 + 4Li+ |
Then LiCoO2 was immediately formed via lithiation of Co3O4. Implying that the kinetics in the LiCoO2 formation in molten LiNO3 might be limited by the oxidation of Co to Co3O4 owing to slow O2 diffusion into bulk Co.
Fig. 3 and 4 show the changes in surface and cross-sectional SEM images of 7 μm-sized Co dots during the growth, respectively. All Co deposits surface in Fig. 4 was covered with a tungsten layer prior to FIB milling for stabilization. The as-deposited Co dots transformed to cylindrical shapes with an aspect ratio of ca. 1.5. Even after heating for 1 h, numerous crystals with platelet shapes spread over the Co surface. Corresponding cross-sectional SEM images clearly show that the individual LiCoO2 crystals grew radially out from the Co dot surface. The length and width of an individual crystal is approximately 1 μm and ∼200 nm, respectively. Notably, three different contrasts were observed in the core. According to corresponding SEM-EDS mapping (Fig. S1†), a dense shell layer of LiCoO2 and/or Li0.6Co0.94O as well as a core of Co and/or Co3O4 were considered to form in the hemispherical assemblies. Furthermore, a nanometer-scale space was formed along the core/shell interface, as shown in Fig. 4(b). Both the platelet crystal growth and dense LiCoO2 shell thickness further increased with the reaction time (Fig. 4(c–e)). All these phenomena implied LiCoO2 formation via Co3O4 strongly associated with the void formation driven by the Kirkendall effect,27–30 which results from the difference between solid-state diffusion rates of the core materials and the rate of O2 diffusion through the shell at the elevated temperatures during oxidation.31–33 We consider that slow O2 diffusion limited LiCoO2 formation and the low solubility of LiCoO2 in molten LiNO3 at the relatively low temperature of 500 °C might create steep gradient of supersaturation toward the surface of Co dots. Such a steep gradient of supersaturation tends to promote the anisotropic crystal growth of LiCoO2 perpendicularly to the Co dot surface driven by kinetic control.9
 | ||
| Fig. 3 Changes in the SEM images of the Co dot directly deposited on Pt substrate: (a) as prepared (b) 500 °C, (c) 500 °C for 1 h (d) 500 °C for 10 h, (e) 500 °C for 50 h. | ||
 | ||
| Fig. 4 Changes in the cross-sectional SEM images of the Co dot directly deposited on Pt substrate: (a) as prepared (b) 500 °C, (c) 500 °C for 1 h (d) 500 °C for 10 h, (e) 500 °C for 50 h. | ||
The plausible formation mechanism based on the above mentioned data is schematically illustrated in Fig. 5. The LiCoO2 phase formation was limited kinetically by oxidation of Co to Co3O4 due to slow O2 diffusion in bulk Co. The LiCoO2 layer were formed immediately from the Co dot surface through the oxidation of Co with resolved O2 and following solid-solution reaction of solid Co3O4 with liquid-phase LiNO3. Because the solubility of LiCoO2 in a molten LiNO3 is low, a small fragment of exposed top part of the LiCoO2 layer was dissolved in the molten LiNO3, leading increased concentration of liquid-phase LiCoO2 around the solid/liquid interface. Simultaneously, the movement of the interface between the diffusion couples of Co3O4 and Co in the core occurred, as the result of the different diffusion rates of Co and O2 at the elevated temperature. Directional material flows resulted from coupled reaction–diffusion phenomena at the solid Co/liquid LiNO3 interface considered to lead to void formation. These voids might be explained by outward transport of fast-moving Co through the top Co3O4 oxide layer and a balancing inward flow of vacancies to the vicinity of the Co3O4 interface. We believe that these two kinetically different reactions created the situation of anisotropic growth of LiCoO2 perpendicularly to the Co dot surface, driven by a kinetically controlled mode. This growth mechanism was strongly similar to that of the growth of the oriented Na2Ti3O7 whiskers from Ti mesh surface using a NaCl flux.34 Once platelet-shaped LiCoO2 crystals were grown from the Co dot surface, the following growth reaction promoted preferential attachment growth along the 〈001〉 directions surrounded by {101} and {104} faces of rhombohedral phased LiCoO2 by kinetically controlled growth.15,35
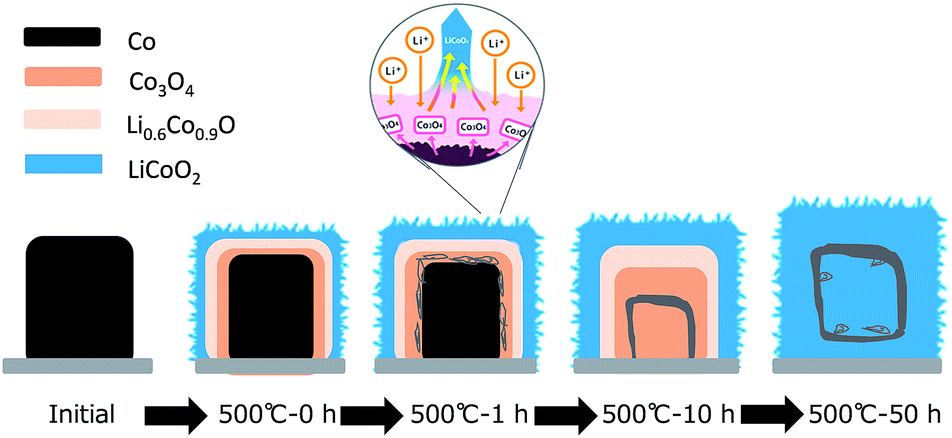 | ||
| Fig. 5 Schematic illustration of the plausible mechanism of the LiCoO2 dot array formation. | ||
Finally, the primitive electrochemical properties of 7 μm-sized LiCoO2 dot arrays as an additive-free LIB-cathode obtained after 10 h of growth was studied. The substrate was heated at 700 °C for 1 h under air to promote crystallization. The a, b faces in the LiCoO2 dot arrays became wide through the contribution of atomic diffusion on the LiCoO2 crystals into the adjacent LiCoO2 crystals at high temperature annealing.15 In addition, the post heating treatment caused a growth of the nanospace formed within the LiCoO2 dot (Fig. S2†). Galvanostatic cycling measurements were conducted using R2032 coin-type cells. No additives for the enhancement of their electron conductivity were used for the studies. Fig. 6 shows the first three cycles of capacity–voltage profiles in the potential range of 2.0–4.2 V vs. Li/Li+, measured at a current density of 50 μA cm−2, corresponding to 0.1C. One C rate indicates the current required to obtain a full charge in 1 h. Excellent reversibility with a high Coulomb efficiency was observed after the second cycle even with no additives, although some irreversible capacity was observed in the first cycle. Owing to the formation of a solid–electrolyte interface (SEI) during the first LIB charging cycle, that the coulombic efficiency seems to be lower; this agrees with trends observed for commercial LiCoO2-based cathodes.24–27 In fact, during the second and subsequent cycles, the coulombic efficiency was recovered to ca. 98%. Therefore, the primitive galvanostatic test demonstrated here revealed that the hollow-structured LiCoO2 crystal array exhibited typical capacity–voltage profiles without any additives. This indicated that electrons were efficiently transferred at the electrode/current collector interface during the intercalation and deintercalation reaction. Meaning that the interfaces provide seamless charge transportation pathways. We further studied the effects of the LiCoO2 dot size on the capacity by comparing the Co dot diameter of 7 μm and 9 μm with almost same coating weight the LiCoO2 dot array, shown in Fig. S3.† The current capacity was noticeably dropped by the increase of the dot size. Thus, we believe such additive-free electrodes with high surface area potentially enhance the specific energy density per electrode volume.
 | ||
| Fig. 6 First 3 charge and discharge curves of the 7 μm-LiCoO2 dot array electrode. | ||
Conclusions
Flux growth of LiCoO2 crystal arrays from the semi-additive-electrodeposited Co dots in molten LiNO3 opens up a new route for the fabrication of microscopically designed LiCoO2 electrodes, which potentially offers significant flexibility in material design and substantially lower cost. It can therefore be responsible for low-cost production of diverse-material LIBs. Time-dependent SEM and XRD studies suggested that the conversion process kinetics involve different reactions, including low LiCoO2 phase formation leading to hollow structures based on the Kirkendall effect and rapid growth of LiCoO2 crystals promoting anisotropic growth. Galvanostatic tests revealed that the hollow-structured LiCoO2 crystal array exhibited typical capacity–voltage profiles with no heavy capacity loss during the first three cycles without any additives. Because the additive-free electrode of LiCoO2 demonstrated here showed similar electrochemical performances of commercially available LiCoO2 based electrodes under the condition of low current density, desirable battery micro-architectures prepared by the combination of semi-additive electrodeposition with flux coating will potentially realize a high energy density and high power density simultaneously owing to additive-free electrodes and the high reactive surface area, respectively. Evaluation of electrochemical characteristics of the additive-free electrodes under a high current density is in progress, and we hope to publish the results in the near feature.Acknowledgements
This work was partially supported by JSPS Grants-in-Aid for Scientific Research (A) (KAKENHI) Grand Number 25249089.Notes and references
- D. E. Bugaris and H.-C. zur Loye, Angew. Chem., Int. Ed., 2012, 51, 3780 CrossRef CAS PubMed.
- D. Elwell and H. J. Scheel, Crystal Growth from High-Temperature Solutions, Academic Press, New York, 1975 Search PubMed.
- S. Oishi, K. Teshima and H. Kondo, J. Am. Chem. Soc., 2004, 126, 4768 CrossRef CAS PubMed.
- K. Teshima, S. Lee, Y. Mizuno, H. Inagaki, M. Hozumi, K. Kohama, K. Yubuta, T. Shishido and S. Oishi, Cryst. Growth Des., 2010, 10, 4471 CAS.
- N. Zettsu, K. Nishikawa, K. Yubuta, K. Sakurai, Y. Yamamoto, Y. Mizuno, S. Oishi and K. Teshima, J. Mater. Chem. A, 2015, 3, 17016 CAS.
- Y. Kim, ACS Appl. Mater. Interfaces, 2012, 4, 2329 CAS.
- Y. Mizuno, N. Zettsu, H. Inagaki, S. Komine, K. Kami, K. Yubuta, H. Wagata, S. Oishi and K. Teshima, CrystEngComm, 2014, 16, 1157 RSC.
- X. J. Yang, W. P. Tang, H. Kanoh and K. Ooi, J. Mater. Chem., 1999, 9, 2683 RSC.
- K. Teshima, H. Inagaki, S. Tanaka, K. Yubuta, M. Hozumi, K. Kohama, T. Shishido and S. Oishi, Cryst. Growth Des., 2011, 11, 4401 CAS.
- N. Zettsu, Y. Mizuno, H. Kojima, K. Yubuta, T. Sakaguchi, T. Saito, H. Wagata, S. Oishi and K. Teshima, Cryst. Growth Des., 2014, 14, 5634 CAS.
- T. Yamada, N. Zettsu, N. Handa, S. Oishi and K. Teshima, Cryst. Growth Des., 2015, 15, 3922 CAS.
- Y. Janssen, D. Santhanagopalan, D. Qian, M. Chi, X. Wang, C. Hoffmann, Y. S. Meng and P. G. Khalifah, Chem. Mater., 2013, 25, 4574 CrossRef CAS.
- J. Li, W. Yao, S. Martin and D. Vaknin, Solid State Ionics, 2008, 179, 2016 CrossRef CAS.
- M. Hirayama, H. Ido, K. Kim, W. Cho, K. Tamura, J. I. Mizuki and R. Kanno, J. Am. Chem. Soc., 2010, 132, 15268 CrossRef CAS PubMed.
- Y. Mizuno, N. Zettsu, K. Yubuta, T. Sakaguchi, T. Saito, H. Wagata, S. Oishi and K. Teshima, Cryst. Growth Des., 2014, 14, 1882 CAS.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863 CrossRef CAS PubMed; Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. S. Eric, A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef PubMed.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651 CrossRef CAS PubMed.
- J. H. Pikul, H. G. Zhang, J. Cho, P. V. Braun and W. P. King, Nat. Commun., 2013, 4, 1732 CrossRef PubMed.
- F. E. Rasmussen, J. T. Ravnkilde, P. T. Tang, O. Hansen and S. Bouwstra, Sens. Actuators, A, 2001, 92, 242 CrossRef CAS.
- A. A. Karimpoor, U. Erb, K. T. Aust and G. Palumbo, Scr. Mater., 2003, 49, 651 CrossRef CAS.
- S. H. Kim, K. T. Aust, U. Erb, F. Gonzalez and G. Palumbo, Scr. Mater., 2003, 48, 1379 CrossRef CAS.
- X. Deng, P. Wei, M. R. Bateni and A. Petric, J. Power Sources, 2006, 160, 1225 CrossRef CAS.
- V. Kublanovsky, O. Bersirova, Y. Yapontseva, H. Cesiulis and E. P. Murphy, Prot. Met., 2009, 45, 588 CAS.
- C.-H. Han, Y.-S. Hong, Y.-S. Park and K. Kim, J. Power Sources, 2001, 92, 95 CrossRef CAS.
- H. Wang, Y. I. Jang, B. Huang, D. R. Sadoway and Y. M. Chiang, J. Electrochem. Soc., 1999, 146, 473–480 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091 CrossRef CAS.
- J. Xie, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda and O. Yamamoto, J. Power Sources, 2009, 189, 365 CrossRef CAS.
- A. D. Smigelskas and E. O. Kirkendall, Trans. AIME, 1947, 171, 130 Search PubMed.
- C. E. Birchenall, J. Electrochem. Soc., 1956, 103, 619 CrossRef CAS.
- J. C. Colson, M. Lambertin and P. Barret, in Proc. 7th Int. Symp. Reactivity of Solids, ed. J. S. Anderson, F. S. Stone and M. W. Roberts, Chapman & Hall, London, 1972, pp. 283–293 Search PubMed.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 30, 711 CrossRef CAS PubMed; S. Mrowec, M. Danlelewski and A. Wojtowicz, J. Mater. Sci., 1998, 33, 2617 CrossRef.
- J. G. Railsback, A. C. Johnston-Peck, J. Wang and J. B. Tracy, ACS Nano, 2010, 4, 1913 CrossRef CAS PubMed.
- J. B. Tracy, D. N. Weiss, D. P. Dinega and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 064404 CrossRef.
- K. Teshima, S. H. Lee, S. Murakoshi, S. Suzuki, M. Kiyohara, K. Yubuta, T. Shishido, M. Endo and S. Oishi, Cryst. Growth Des., 2010, 10, 2533 CAS.
- K. Takahashi, S. J. Limmer, Y. Wang and G. Z. Cao, Jpn. J. Appl. Phys., 2005, 44, 662 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17459g |
| This journal is © The Royal Society of Chemistry 2015 |