
Functionalized Fe3O4 nanoparticles: influence of ligand addition sequence and pH during their continuous hydrothermal synthesis†
 a
and
a
and
Abstract
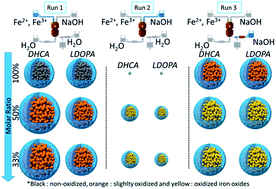
In this study we report various new efficient ways to synthesize and modify in situ magnetite (Fe3O4) iron oxide nanoparticles (NPs). Thanks to an apparatus especially developed for this new method of grafting, the NPs have been synthesized and functionalized by 3,4-dihydroxyhydrocinnamic acid (DHCA) or 3,4-dihydroxy-L-phenylalanine (LDOPA) in one step and under hydrothermal conditions using varying concentration ratios ([organic molecules]/[ferrous and ferric ions]). The organic molecules were added before or after the NP synthesis. The addition of these organic molecules modifies the structure, the morphology, the oxidation degree and the growth of the crystallites. Adding the organic molecules before the synthesis step and under acidic conditions increases the average crystallite size and prevents further oxidation whereas under basic conditions the growth is stopped but a partial oxidation of magnetite to maghemite NPs is observed. Adding DHCA or LDOPA after the synthesis step results in a modification of the lattice structure and oxidation degree of the NPs but does not change the average size. This study underlines the importance of the sequence of the addition of organic molecules on the synthesis of NPs.
 Please wait while we load your content...
Please wait while we load your content...

