
d-Glucose based synthesis of proline–serine C–C linked central and right hand core of a kaitocephalin-a glutamate receptor antagonist†
Abstract
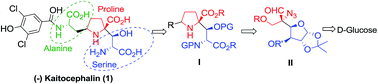
The synthesis of the proline–serine core of kaitocephalin 1 was accomplished starting from D-glucose wherein, C2 of D-glucose provided the carboxylic acid functionality of serine; while the amino- and β-hydroxy groups of the serine fragment were amenable from the C3 and C4 hydroxy groups of sugar. The key intermediate to construct substituted proline with the appropriate quaternary carbon framework of the target molecule was constructed using the Jocic–Reeve and Corey–Link reaction sequence with the desired stereochemistry from the C5-centre of D-glucose.
 Please wait while we load your content...
Please wait while we load your content...

