
Novel phosphorescent iridium(iii) complexes containing 2-thienyl quinazoline ligands: synthesis, photophysical properties and theoretical calculations†
Abstract
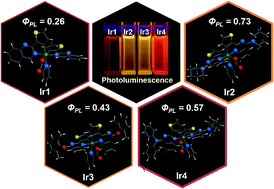
The easy tailoring of organic ligands of iridium(III) complexes provides a facile way to tune their opto-electronic properties for applications in high efficiency phosphorescent light emitting diodes. Herein, a series of yellow and red emitting phosphorescent iridium complexes based on 2-thienyl quinazoline derivatives are successfully synthesized and systematically characterized with various opto-electronic properties. The X-ray crystal structures demonstrate that the iridium centers in the complexes with bulky substituents on the 4-position of quinazolyl rings prefer to chelate with the N atoms in the 1-position of quinazolyl rings. Both experiment and theoretical studies indicate that the steric hindrance along with the electron-donating effect of substituents on the C^N ligands enhances the emission quantum yields, accompanied by significant emission shifts. Two yellow phosphorescent iridium complexes (Ir2 and Ir3) are successfully designed and exhibit moderate emission efficiencies, through the incorporation of bulky ligands with strong electron-donating abilities (piperidine for Ir2 and 2,6-dimethyl-phenoxy for Ir3, respectively). The synergistic effect of electron structure and hindrance of ligand is believed to be a promising strategy for tuning the opto-electronic properties of iridium complexes.
 Please wait while we load your content...
Please wait while we load your content...

