
Biocompatible locust bean gum mesoporous matrices prepared by ionic liquids and a scCO2 sustainable system†
Márcia G. Ventura*,
Ana I. Paninho,
Ana V. M. Nunes,
Isabel M. Fonseca and
Luís C. Branco*
LAQV-REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Monte de Caparica, Portugal. E-mail: mm.ventura@fct.unl.pt; l.branco@fct.unl.pt; Fax: +351 212948550; Tel: +351 212948300
First published on 7th December 2015
Abstract
Locust bean gum (LBG) is a natural polymer that belongs to the increasingly attractive biodegradable polymers derived from natural sources. This polymer possesses a number of appealing features such as non-toxicity that confer it a high potential for the use in fields such as pharmacy and food industries. In this work, three methylimidazolium-based ionic liquids (ILs) were used as alternative media in the preparation of biocompatible LBG gel matrices. The process consists of dissolving the biomaterial in a selected IL followed by a multi-step solvent exchange using a mixture of water and ethanol. The final gel (internal surface areas ranging from 60 to 180 m2 g−1) is then dried using a semi-continuous high pressure CO2 extraction process. To the best of our knowledge this is the first time that LBG mesoporous gel matrices are reported using this efficient sustainable method.
Introduction
Natural polymers extracted either from plants and animals are regarded as promising ingredients for the production of bio inspired materials used as carriers or adsorbents that could replace the synthetic ones. In this context, the natural polymers exhibit some characteristics which make them very attractive. They are economical, easily available, non-toxic, biodegradable and, with few exceptions biocompatible.1,2 Locust bean gum is a natural polymer obtained from the endosperm of the seed of the carob (locust) tree, Ceratonia siliqua which is abundant in the entire Mediterranean area. It is a non-starch polysaccharide consisting mainly of a neutral galactomannan polymer made up of linear mannose chains with an unit of D-galactose linked to the main backbone by α-glycosidic bonds every fourth or fifth chain units.3,4 The main chain of LBG is structurally similar to that of cellulose which is completely insoluble in water as a result of chain association through hydrogen bonding. In LBG, the substitution of the mannose (M) chain by galactose (G) units prevents extensive chain association and improves the solubility of the polymer in water. Having a high ratio M/G (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the LBG presents a limited solubility in cold water with propensity to form aggregates. This polysaccharide requires heating process (T > 85 °C during 10 min) to achieve full hydration, solubilization and maximum viscosity.4,5 However, it presents a high gelling capacity which, among other properties, makes it very appealing in particular for biomedical and pharmaceutical applications. Improving the gelling capacity of this polymer is usually done through the association of two different polysaccharides exhibiting a stronger synergy.4 In case of LBG, the most effective synergies are established with xanthan gum and carrageenan4,5 resulting in the formation of strong and self-sustaining gels. Several studies on the LBG interaction with xanthan and carrageenan6–10 have been reported in several fields of application, from oral, buccal and colonic delivery to topical applications. In these studies, the mixture of LBG and a second polysaccharide is optimized in order to improve the properties of the gel from a desired formulation. Another way to obtain LBG consistent gel matrices is through the use of a cross-linker such as glutaraldehyde promoting the formation or enhancement of the chemical gel networks.11
:1), the LBG presents a limited solubility in cold water with propensity to form aggregates. This polysaccharide requires heating process (T > 85 °C during 10 min) to achieve full hydration, solubilization and maximum viscosity.4,5 However, it presents a high gelling capacity which, among other properties, makes it very appealing in particular for biomedical and pharmaceutical applications. Improving the gelling capacity of this polymer is usually done through the association of two different polysaccharides exhibiting a stronger synergy.4 In case of LBG, the most effective synergies are established with xanthan gum and carrageenan4,5 resulting in the formation of strong and self-sustaining gels. Several studies on the LBG interaction with xanthan and carrageenan6–10 have been reported in several fields of application, from oral, buccal and colonic delivery to topical applications. In these studies, the mixture of LBG and a second polysaccharide is optimized in order to improve the properties of the gel from a desired formulation. Another way to obtain LBG consistent gel matrices is through the use of a cross-linker such as glutaraldehyde promoting the formation or enhancement of the chemical gel networks.11
In the last decade, ILs have emerged as a new class of efficient solvents for the dissolution of starch polysaccharides such as cellulose and related lignocellulosic materials.11–15 These solvents opened up a broad variety of new opportunities on the chemistry of these materials since they are able to regenerate the biopolymers into fibbers, films and beads and are particularly useful on their homogeneous chemical modification.11 In a general way, when a polysaccharide is dissolved into an ionic liquid, the hydrogen bonds within the polymer are broken and new ones can be established and a reorganization of previous arrangement can occur. ILs appear to be particularly effective on this process due to their high ionic character.11 ILs are defined as organic salts with low melting temperatures (<100 °C) among which the most promising for the biopolymer approaches have been based on 1-alkyl-3-methylimidazolium cation structure as well as some good examples with ammonium and pyridinium cations.12 In turn, the anions which have proved to be the most promising are chloride, acetate and formate. The mechanism is still not well known but apparently both cations and anions contribute on the dissolution process through either specific (hydrogen bonds) or non-specific interactions.11 Although in the case of LBG, hydrocolloid, the dissolution question has not the same relevance as in the case of cellulosic materials, applying ILs on the dissolution of this polymer brings about the possibility of improving its processability and of obtaining gels with different chemical structures and/or physical properties to be used accordingly to the desired application. Aerogel technology is an effective way of obtaining ultralight and highly porous polymeric materials which are very promising for delivering systems.15–17 Gels with such characteristics can be produced from a solution of a polymer which forms a gel network by entrapping the original solvent. The solvent within the gel network can be then replaced by air through supercritical drying without the collapsing of the gel structure.18 Typical values of porosity, density and surface area for these systems are 90–99%, 0.07–0.46 g cm−3 and 70–680 m2 g−1 respectively, which make them highly porous drug delivery systems with a high surface area associated.16 There are some reports using several polysaccharides for the production of ultralight and porous gels19–23 but none was found for the LBG. Herein, LBG is used for the production of ultralight and mesoporous gels through supercritical CO2 extraction and using both water and different ILs as dissolution media of the biopolymer. ILs were selected based on methylimidazolium cation combined with different anions. According with type of selected ILs for LBG dissolution, it is possible to evaluate how the IL structure influences the textural properties of the final gel. Once the LBG is water soluble, the characteristics of the gels produced through dissolution into ionic liquids were also compared with those obtained for the gel dissolved in water in similar conditions. Different parameters were also evaluated such as temperature, reaction time and biopolymer to solvent mass ratio used during the dissolution process, in order to adjust the properties of the final gel. An attempt on the improvement of the textural properties of the final gel by varying the composition of the solvent mixture used during the supercritical drying process was made for one of the produced samples.
Results and discussion
As hydrocolloid (as indicated in Fig. 1), LBG can be dissolved in water giving a gel. Its dissolution in ILs was attempted in order to verify if a change and/or improvement of textural properties associated with the gel formed by the biopolymer is achieved. The solubilization of LBG and the gelation of the homogeneous liquid solutions were observed for each selected IL as well as using water. A stable gel was achieved by dissolution into 1-butyl-3-methylimidazolium chloride [bmim][Cl] after 4 h at 80 °C. Higher temperature and extended reaction time were required for the ILs 1-ethyl-3-methylimidazolium acetate [emim][Ac] and 1-(2-hydroxyethyl)-3-methylimidazolium chloride [C2OHmim][Cl] in order to obtain a stable gel (see Table 1). Similar values for the dissolution time and temperature were used for the production of ion gels from different polysaccharides including neutral polysaccharides, such as the galactomannans, the authors have fabricated ion gels of LBG, guar gum and fenugreek gum using [bmim][Cl] and a dissolution temperature of 100 °C during 5 h.24,25 The gel produced through the dissolution in [C2OHmim][Cl] showed a stable structure but the absence of a regular shape suggests a more randomized gel network formation. In fact, the presence of hydroxyl group as substituent of methylimidazolium cation must influence the dissolution as well as the network reorganization during precipitation through the formation hydrogen bonds with the polymer. In case of [bmim][Cl] and [emim][Ac] no substituent groups are able to promote hydrogen bonds. Consequently, a more organized net structure seems to be formed and the respective gels assume a regular shape. This factor can be crucial for stability and strength of the gel. More organized structures allow the formation of stronger gels which are more able to be formed using lower reaction times and polymer mass. In this context, recent publications about dissolution of other polysaccharides reported that [bmim][Cl] is the more efficient on this dissolution process giving origin to stronger gels at lower reaction times and temperatures.11,12 Despite a lower biopolymer mass was used (5% w/w), samples produced through the dissolution in water present a more compact macroscopic behaviour but a gel was obtained in all the used conditions (Table 1). The mass ratio of 10% (w/w) was attempted using water as solvent but the complete dissolution of the LBG was not achieved in this case.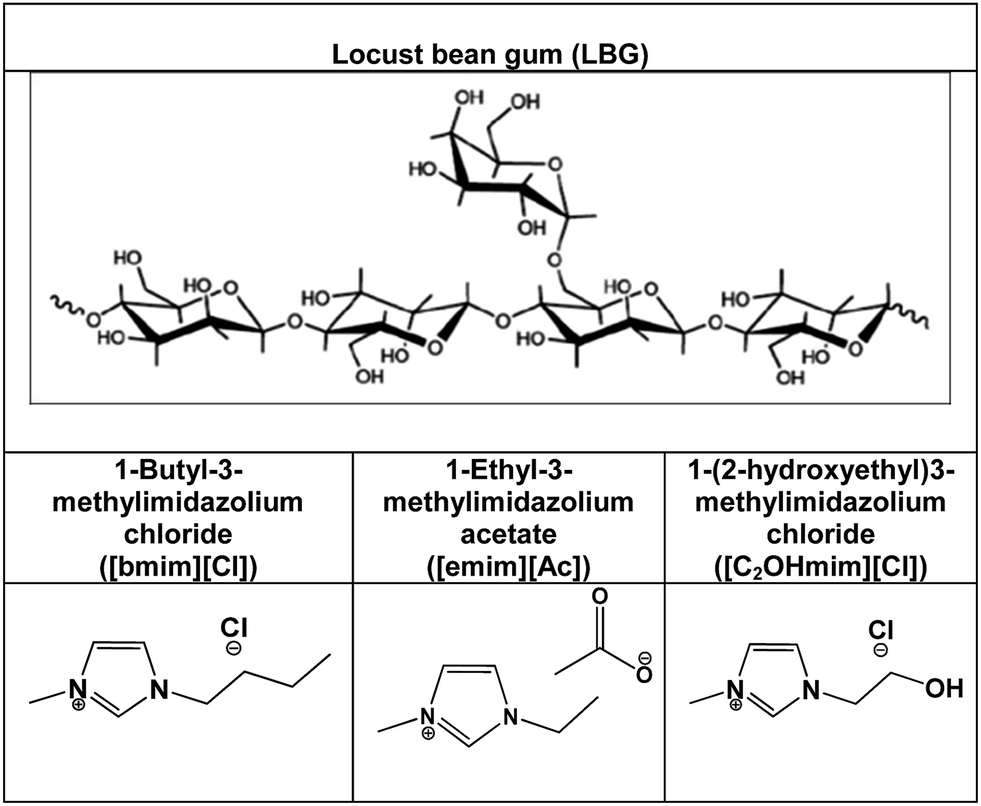 | ||
| Fig. 1 Chemical structures of the LBG and selected ILs. | ||
| Solvent | No. | Locust bean gum (g) in 1 g of solvent | Dissolution temperature (°C) | Dissolution time (h) | Sample appearance in 100% ethanol |
|---|---|---|---|---|---|
| [bmim][Cl] | 1 | 0.1 | 80 | 4 | Gel |
| 2 | 0.1 | 80 | 2 | Weak gel | |
| 3 | 0.1 | 60 | 4 | Gel | |
| 4 | 0.025 | 80 | 4 | Weak gel | |
| 5 | 0.025 | 80 | 2 | Weak gel | |
| [emim][Ac] | 6 | 0.1 | 100 | 6 | Gel |
| 7 | 0.1 | 100 | 4 | No gel | |
| 8 | 0.025 | 100 | 6 | No gel | |
| [C2OHmim][Cl] | 9 | 0.1 | 100 | 6 | Gel (no regular shape) |
| 10 | 0.1 | 100 | 4 | No gel | |
| 11 | 0.025 | 100 | 6 | Very weak gel | |
| Water | 12 | 0.05 | 90 | 4 | Gel |
| 13 | 0.05 | 90 | 2 | Gel | |
| 14 | 0.05 | 60 | 4 | Gel | |
| 15 | 0.025 | 90 | 4 | Gel | |
| 16 | 0.025 | 90 | 2 | Gel |
The obtained biopolymer solution was immersed in an aqueous bath immediately after the dissolution step. The concentration of ethanol in the aqueous bath changed between 10 to 90%, being the last bath constituted uniquely by ethanol. Images from the resulting alcogels using different ILs and water can be observed on Fig. 2.
 | ||
| Fig. 2 Alcogel and gel pictures and the SEM images for the surface and cross section of the gels produced through the dissolution into [bmim][Cl] (first row), [emim][Ac] (second row), [C2OHmim][Cl] (third row) and water (fourth row). | ||
The microstructures of the gels achieved after the supercritical CO2 drying of the mentioned alcogels were obtained through SEM as also presented in Fig. 2. All samples display the characteristic open-pore structures independently of the solvent in which the biopolymer was dissolved. According to images the pore distribution is very wide, from a few nanometers to several micrometers.
The gel materials produced through the dissolution into the ILs showed an uniform, opaque and white surface and were compressible despite the initial volume was not recovered after a compressive action. The gel produced through the dissolution into water presented a higher hardness. The weak gels resulting from the regeneration bath (samples 2, 4, 5 and 11 – see Table 1) gave very hard and thin structures after supercritical CO2 drying suggesting a low or absent porosimetry as a consequence of the destruction of the possibly weak network that was formed during the dissolution process. The sample 3 produced through the dissolution into [bmim][Cl] at lower temperature (60 °C) became a powder after supercritical CO2 drying meaning that at this temperature the formation of an enough strong and structured network was not assured. All the produced gels are partially re-dissolved into water.
The TGA studies were performed in order to evaluate the final thermal stability of each gel as well as possible presence of IL in the gels after supercritical CO2 drying. Although all ionic liquids used in this work are completely soluble in ethanol, a residual amount of IL could interfere with the porosity and specific surface area of the LBG gel. Furthermore, ILs are mostly not soluble in scCO2, for what they will not be removed in the drying step, in case the washing step fails. The TGA curves on Fig. 3 showed the thermal profile for the LBG gels produced by dissolution into the three selected ILs and water. In general, the LBG gels produced by dissolution in ILs showed higher thermal stability than gel using water. The differential scanning calorimetry (DSC) curves (see ESI Fig. 1S†) indicated us at least two significant thermal phenomena for all the produced gels. The curves obtained when ILs were used as solvent present a similar behavior to the one obtained for water. The first stage between 60 and 120 °C may be attributed to water elimination which is retained on the samples. The weight loss in this step varies from 10 to 15% for the analyzed samples. The second stage at 280–320 °C is related to the polysaccharide decomposition.26
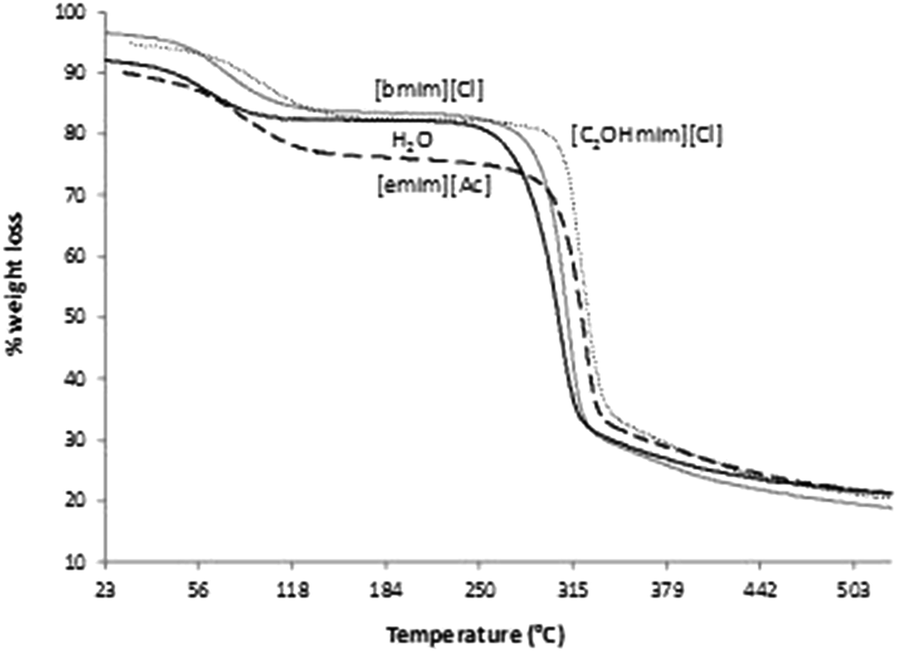 | ||
| Fig. 3 TGA thermograms of LBG gels produced through the dissolution into the ILs [bmim][Cl] solid gray line, [emim][Ac] dashed line, [C2OHmim][Cl] dotted line and water solid black line. | ||
The gels suffered a weight loss between 45–55% at temperature near 320 °C due to LBG thermal decomposition. The values obtained in this work are in conformity with the values reported by Martins et al.27 for LBG films, with values ranging between 230 and 330 °C. Given the similarities obtained in curves from gels produced through the dissolution into ILs and the one dissolved into water we have assumed that residual ILs could eventually exist only on trace amounts and hence are not interfering with the porosity values of the samples.
Table 2 summarizes the characterization data obtained through N2 porosimetry for the gel samples. According to the results, the average pore diameter is on the mesopore size range and is similar for the samples dissolved in the different ILs and water despite on this later situation pores are in average smaller. Low BET surface areas are presented for samples 13 and 16 in which shorter dissolution times were tested. Lower surface areas can be explained by poor reticulation which might be a consequence of the short dissolution reaction times. Contrarily, sample 9 (dissolution was made in [C2OHmim][Cl]) showed a higher surface area corresponding to a higher microporosity, fact that can be observed on Fig. 2. The possibility of the establishment of additional hydrogen bonds between the hydroxyl group at terminal substituted group in the imidazolium cation and the chain of the biopolymer should be relevant for higher number of formed micropores. Comparing the values for the t-plot of micropore volume, similar values were determined on samples 1 and 6, respectively 6.63 × 10−3 cm3 g−1 and 5.12 × 10−3 cm3 g−1, while in case of sample 9 it is higher, 18.6 × 10−3 cm3 g−1 putting in evidence the higher microporosity and much lower in case of sample 12, 0.751 × 10−3 cm3 g−1.
| Sample no. | BET area (m2 g−1) | BJH-adsorption average pore diameter (nm) | BJH-desorption average pore diameter (nm) |
|---|---|---|---|
| 1 | 65.6 | 17.2 | 16.4 |
| 6 | 65.9 | 18.9 | 16.8 |
| 9 | 184.8 | 16.6 | 20.1 |
| 12 | 66.7 | 13.3 | 9.8 |
| 13 | 0.5 | 2.5 | 43.3 |
| 14 | 97.2 | 11.9 | 8.6 |
| 15 | 60.0 | 15.9 | 14.4 |
| 16 | 21.4 | 14.4 | 29.5 |
The values of average pore diameter presented on Table 2 are similar to the ones determined by other authors for biopolymers such as starch, alginate, k-carrageenan, cellulose, chitosan and chitin despite generally lower surface areas were obtained.16
Bulk density determined through mercury porosimetry ranges from 0.15 g cm−3 to 0.41 g cm−3 with an exception for sample 13 with a bulk density determined as 0.71 g cm−3. The lowest values, 0.15 g cm−3 and 0.20 g cm−3 were obtained for the lower used mass ratios (0.025) in samples 15 and 16 respectively. Except for sample 13, the determined values are in agreement with values presented for other polysaccharides gels.16
In order to obtain the macroporosity of the produced matrices, mercury porosimetry was performed. The pore size distribution determined through mercury porosimetry of the gels produced from dissolution into [bmim][Cl], [emim][Ac] and water is presented on the ESI (Fig. 2S†). The results for sample 9 dissolved into [C2OHmim][Cl] are absent due to its destruction during the analysis (the sample was too compressed after mercury intrusion). The three obtained distributions are Gaussian with a large range of pores sizes from mesopores (2–50 nm) to macropores (>50 nm). A higher incidence on pores with diameters larger than 100 nm is observed for samples 1 and 12. Due to the compressibility of these samples, it is difficult to assure that the selected intrusion volume of mercury is returning an accurate profile of the macropores size, nevertheless, from the observation of the SEM images for these samples, pore diameters from 200 nm to 3000 nm are rather observed.
The textural properties of the final gel, namely surface area and pore diameters can be significantly affected by the used binary mixture (CO2 + solvent) composition at the beginning of supercritical extraction process.28 This observation results from a combined effect of solvent degree of swelling/mixture density that, at this stage, due to gel high physical flexibility, is able to control the matrix pores. Ethyl lactate has shown promising results in this field and thus a study on the effect of this solvent during supercritical extraction over the porosity of samples produced through the dissolution into [bmim][Cl] was attempted. The SEM image (Fig. 4) reveals an gel sample with areas containing highly expanded pores with diameters close to 8000 nm and higher than the ones observed in sample 1 produced in the same way but dried with a mixture (CO2 + ethanol). Apparently, associated to the areas with higher macroporosity there are also areas of extended microporosity. Different physico-chemical properties between ethyl lactate and ethanol lead to different interactions between the solvent and the matrix which allows to obtain final different gel morphologies [AN]. In this case ethyl lactate higher density and almost double viscosity in comparison with ethanol, are likely inducing an increase in the size of the pores. The results obtained from N2 porosimetry revealed a higher surface area (BET area of 167.0 m2 g−1) when compared with the surface area of sample 1. The total volume associated to the microporous was 14.5 × 10−3 cm3 g−1 and the average pore diameter obtained from BJH adsorption and desorption was estimated in 14.8 and 13.0 nm, respectively. The pore size distribution obtained from mercury porosimetry for this sample reveals a more homogeneous distribution with a higher incidence in macroporous by comparing with the one obtained for sample 1 (Fig. 2S on ESI†). The performance of the LGB gel for drug delivery applications was assessed through ibuprofen impregnation with CO2 and posteriorly kinetic release studies. Ibuprofen impregnation into LBG gel was performed in the same apparatus used for gel drying, but in a batch mode with some modifications. The reactor was loaded with ibuprofen plus a magnetic stir bar at the bottom and packed with a net metallic disk and filter paper at the top, where the gel was placed. The reactor was then immersed in the thermostated water bath and the drug allowed to impregnate into the matrix for six hours at 313.2 K and 14 MPa.
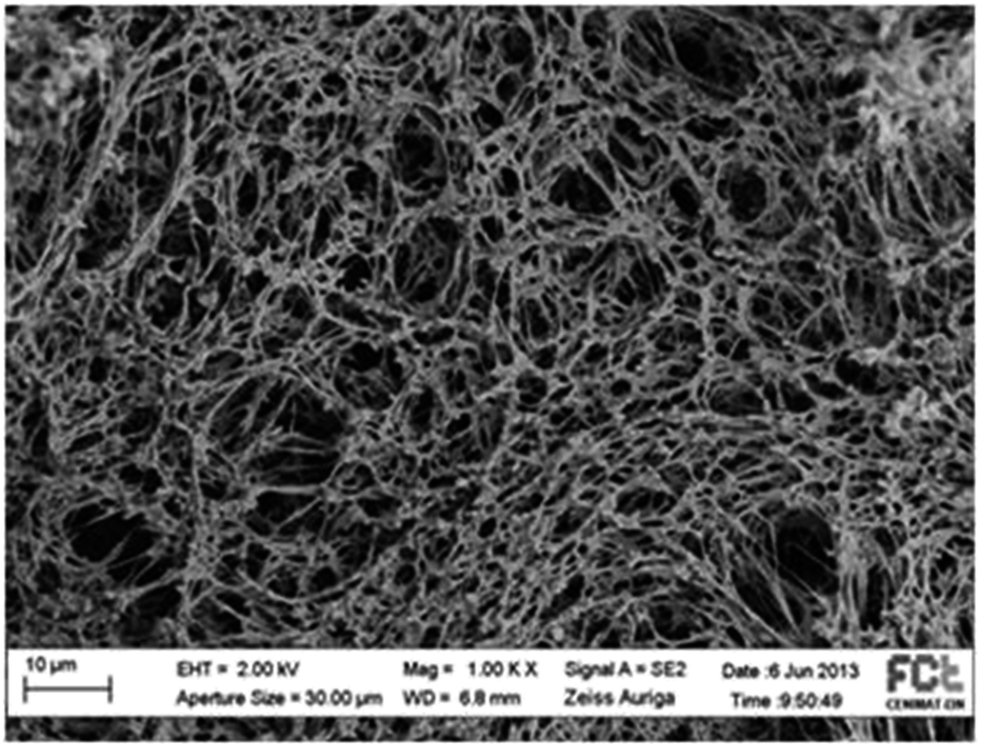 | ||
| Fig. 4 SEM image for the cross section of the gel produced through the dissolution into [bmim][Cl] and dried with a mixture (CO2 + ethyl lactate). | ||
The amount of ibuprofen impregnated per gram of sample was determined to be 84.5 ± 5 mg.
The release rate of ibuprofen from the LBG gel was followed by the dispersed amount method, in pH 7.4 phosphate buffered saline (PBS) solution. The release profile obtained is presented in Fig. 3S on the ESI.† Around 63% (±2%) of ibuprofen was released after 60 min and 91% (±2%) was reached after 120 min which is close to the amount released (96 ± 1%) at 180 min. Closer values for the controlled release of ibuprofen were obtained for matrices such as polymeric microgels of sodium alginate and acrylic acid.29,30 For these materials it was reported that a 70% drug release was reached after 6 h and between 85 and 100% after 12 h.31,32
Conclusions
Pure LBG mesoporous matrices were produced through the dissolution of the biopolymer into three different ILs and into water. The gel materials produced in this sustainable process are opaque, white in colour and have a good compressibility. The one produced through the dissolution into water presented a higher hardness and lower thermal stability. The integrity of the produced gels/gels depends on the mass of polymer, reaction time and selected temperature during the dissolution process. The IL [bmim][Cl] showed higher efficiency, dissolving the same amount of biopolymer in less time and at lower temperature. The porosity obtained whenever an IL was used is higher when compared to the one obtained for the samples produced through the dissolution into water. Improving the surface area of the gel is possible by changing the composition of solvent mixture during the supercritical CO2 drying. For this purpose, ethyl lactate can be efficiently used instead ethanol. Facing the obtained results, supercritical CO2 technology has proven to be efficiently applied on these matrices with the additional possibility of a more effective control over its porosity. More experiments using different supercritical drying mixture compositions will be required in order to determine in which extent the porosimetry of the materials can be improved.These innovative materials consisting entirely of LBG will be suitable to be used mainly in the pharmaceutical and food industries. The LBG gel matrices might be a more efficient alternative to the existing pharmaceutical formulations constituting the basis for new promising functional materials in this field. On the other hand, and beyond the already existent roles on food industry as thickening and structuring agent these matrices can be applied both as encapsulation and delivery systems for ingredients such as vitamins, antioxidants, flavoring and colorants. Despite the promising results more research will be needed in order to determine its performance in the different considered fields.
Experimental section
Materials and methods
The LBG used in these work is for food purposes and was purchased at Sosa ingredients catalogue.The selected ILs [bmim][Cl], [emim][Ac] and [C2OHmim][Cl] were acquired from Solchemar Lda (purity higher than 99%). Ethanol and ethyl lactate from Sigma-Aldrich as p.a. grade (99% of purity). DSC and TGA studies as well as SEM analysis were performed in CENIMAT (FCT/UNL). Mercury porosimetry and N2 adsorption–desorption analysis were done in REQUIMTE (FCT/UNL).
Preparation of solutions and gels
The dissolution, gelling and gel formation properties of LBG were studied in three different ILs and in water. For this purpose the amount of used polymer and its dissolution time into the solvent were the tested parameters. The selected ILs [bmim][Cl], [emim][Ac] and [C2OHmim][Cl] are all derived from methylimidazolium cation unit allowing to evaluate the effect of substituent chain (n-alkyl or alcohol) of the cation as well as the type of anion on the dissolution capacity. The dissolution of the polymer into water was performed on same conditions to those used with ILs and results compared.The gels were prepared by the dissolution into ILs according to a typical preparation procedure.15 The polymer (1–10% w/w) is mixed with the IL in flask immersed in an oil bath with stirring at a given temperature. The time and temperature of heating was changed in order to optimize the dissolution process for the different used concentrations of the polymer and ILs. Dissolution times up to 6 hours and temperatures of reaction up to 100 °C were used.
The solution obtained from the dissolution step is then introduced into a regeneration bath consisting of a room temperature aqueous solution containing either 10%, 50% or 90% ethanol. The immersion time in each bath is at least during one night and for each ethanol concentration the bath is replenished three times. The alcogel is then obtained using one last bath composed by pure ethanol.
Drying in supercritical conditions
Supercritical fluid extraction of the alcogel was carried out using a semi-continuous high pressure apparatus, described in detail elsewhere.28 Briefly, the high pressure apparatus is composed of a cylindrical extractor of 12 cm3, positioned vertically in a thermostated water bath, heated to the desired temperature by means of a controller that maintained the temperature within ±1 K (JP Selecta Termotronic). The alcogels, were placed at the bottom of the extractor always immersed in 5 ml of ethanol in order to avoid the risk of solvent evaporation from the gel, before being in contact with CO2 atmosphere. The addition of CO2 was made using a pneumatic compressor (Electrolux) connected to the top of the reactor, through a 1/16 stainless steel tubing, which passes through the sealing system, releasing the CO2 directly at the bottom of the reactor. The pressure in the system was measured with a pressure transducer Setra Datum 2000 TM calibrated between 0 and 34.3 MPa (with a precision of ±0.1% at the lowest pressure). CO2 exit the system through two high pressure valves, connected directly to the sealing system at the top of the extractor, and further connected to a gas flow meter. The two high pressure valves are slowly released and manipulated to control the CO2 flow and maintain constant the pressure inside the system.The process was carried out during two hours, with a constant CO2 flow rate of 3 g min−1. A temperature of 313.2 K and a pressure of 15 MPa were selected to assure supercritical conditions of the binary system (ethanol + CO2). Finally, an experiment in which ethanol was replaced by ethyl lactate was performed, to determine the solvent influence on gel final morphological properties.
Characterization of the solids
Textural properties of the LBG gels were determined by low temperature N2 adsorption–desorption analysis. Prior to the measurements, samples are dried under vacuum (<1 mPa) at 60 °C at least for 12 h. The adsorption isotherms where obtained at 77 K. Specific surface area is determined by the BET (Brunauer–Emmett–Teller) method. Pore volume diameter is estimated using the BJH (Berrett–Joyner–Halenda) method. The mercury porosimetry was performed after weighing samples through the element intrusion until 30000 psi.
Micrographs of the samples were recorded through scanning electron microscopy (SEM) using a Zeiss Auriga microscope operating at 1 kV. Gold was used for the sputtering of the samples in order to minimise charging and improve the image contrast.
Thermal stability of samples was analysed by thermogravimetric analyses (TGA). Samples were placed in a balance system and heated from 20 °C to 550 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.
Impregnation and release case study
The impregnation of ibuprofen on the LBG matrix produced using ethyl lactate was performed adopting a semi-continuous strategy on the same high pressure apparatus. The approach was to sequentially perform the impregnation and drying of the gel in a one-pot process. The high pressure apparatus was adapted for the experiment: an alternative CO2 line with a tubular reactor enclosing the drug was assembled before the extractor. The CO2 flow was forced to pass through the tubular reactor in order to saturate with the drug. The saturated CO2 flow pass through the cylindrical extractor and the drug adsorb on the gel matrix. In this way the impregnation can be performed following the drying process. The mass of ibuprofen used on the impregnation process was 345 mg.The ibuprofen content in the gel matrix was quantified spectrophotometrically. To extract the drug from the gel matrix for drug content quantification, drug-loaded gel was transferred to a phosphate buffer solution with pH 6.8 and incubated for 2 h for swelling. The swelling gels were then broken up and the obtained solution centrifuged. The ibuprofen content was thus determined measuring its absorption at 223 nm.
The release experiment was carried out through the immersion of the drug loaded gel matrix in a determined volume of phosphate buffer at pH 7.4 at 37 °C. While the dissolution medium was gently agitated, aliquots of dissolution medium (5 ml) were withdrawn manually after 5, 10, 15, 20, 25, 30, 60, 120 and 180 min and replaced with an equal amount of fresh dissolution medium to maintain sink conditions. The dissolution sample was then analyzed for drug content using UV-vis spectrophotometry.
Acknowledgements
FCT-MCTES (Portugal) is acknowledged for financial support through the projects PTDC/CTM/103664/2008, PTDC/CTM-NAN/120658/2010, PTDC/EQU-EQU/104552/2008, and post-doc grants SFRH/BPD/40008/2007 (MGV) and SFRH/BPD/74994/2010 (AVMN). The authors would like to thanks to Dr Nuno Costa for the technical support on the porosimetry analysis.Notes and references
- V. S. Kulkarni, K. D. Butte and S. S. Rathod, Int. J. Res. Pharm. Biomed. Sci., 2012, 3, 1597 Search PubMed.
- A. M. Avachat, R. R. Dash and S. N. Shrotriya, Indian Journal of Pharmaceutical Education and Research, 2011, 45, 86 Search PubMed.
- V. D. Prajapati, G. K. Jani, N. G. Moradiya, N. P. Randeria and B. J. Nagar, Carbohydr. Polym., 2013, 94, 814 CrossRef CAS PubMed.
- G. O. Phillips and P. A. Williams, in Handbook of Hydrocolloids, Woodhead Publishing Limited, England, 2000, p. 137 Search PubMed.
- M. Dionísio and A. Grenha, J. Pharm. BioAllied Sci., 2012, 4, 175 CrossRef PubMed.
- J. Sujja-areevath, D. Munday, P. Cox and K. Khan, Eur. J. Pharm. Sci., 1998, 6, 207 CrossRef CAS PubMed.
- S. Hirsh, V. Binder, V. Schehlmann, K. Kolter and K. H. Bauer, Eur. J. Pharm. Biopharm., 1999, 47, 61 CrossRef.
- M. Yamagar, V. Kadam and R. Hirlekar, Int. J. Res. Pharm. Biomed. Sci., 2004, 2, 453 Search PubMed.
- C. W. Vendruscolo, I. F. Andreazza, J. M. L. S. Ganter, C. Ferrero and T. M. B. Bresolin, Int. J. Pharm., 2005, 296, 1 CrossRef CAS PubMed.
- C. Vijayaraghavan, S. Vasanthakumar and A. Ramakrishanan, Pharmazie, 2008, 5, 342 Search PubMed.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712 CrossRef CAS PubMed.
- T. Liebert and T. Heinze, BioResources, 2008, 3, 576 Search PubMed.
- I. Kilpelainen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142 CrossRef PubMed.
- A. Biswas, R. L. Shogren, D. G. Stevenson, J. L. Willett and P. K. Bhowmik, Carbohydr. Polym., 2006, 66, 546 CrossRef CAS.
- O. Aaltonen and O. Jauhiainen, Carbohydr. Polym., 2009, 75, 125 CrossRef CAS.
- C. A. Garcia-González, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2011, 86, 1425 CrossRef.
- I. Smirnova, J. Mamic and W. Arlt, Langmuir, 2003, 19, 8521 CrossRef CAS.
- J. Fricke, Sci. Am., 1988, 258, 92 CrossRef CAS.
- R. R. Escudero, M. Robitzer, F. Di Renzo and F. Quignard, Carbohydr. Polym., 2009, 75, 52 CrossRef CAS.
- J. Innerlohinger, H. K. Weber and G. Kraft, Macromol. Symp., 2006, 244, 126 CrossRef CAS.
- R. J. White, V. L. Budarin and J. H. Clark, ChemSusChem, 2008, 1, 408 CrossRef CAS PubMed.
- F. Quignard, R. Valentinw and F. Di Renzo, New J. Chem., 2008, 32, 1300 RSC.
- M. Robitzer, F. D. Renzo and F. Quignard, Microporous Mesoporous Mater., 2011, 140, 9 CrossRef CAS.
- J. I. Kadokawa, J. Biobased Mater. Bioenergy, 2013, 7, 3–11 CrossRef CAS.
- A. Takada and J. I. Kadokawa, Biomolecules, 2015, 5, 244–262 CrossRef CAS PubMed.
- M. J. Zohuriaan and F. Shokrolahi, Polym. Test., 2004, 23, 575 CrossRef CAS.
- J. T. Martins, M. A. Cerqueira, A. I. Bourbon, A. C. Pinheiro, B. W. S. Souza and A. A. Vicente, Food Hydrocolloids, 2012, 29, 280 CrossRef CAS.
- A. B. Paninho, C. Barbosa, I. D. Nogueira, V. Najdanovic-Visak and A. V. M. Nunes, J. Supercrit. Fluids, 2013, 83, 35 CrossRef CAS.
- J. Andersson, J. Rosenholm, S. Areva and M. Linde, Chem. Mater., 2004, 16, 4160 CrossRef CAS.
- E. Santamaría, A. Maestro, M. Porras, J. M. Gutiérrez and C. González, J. Solid State Chem., 2014, 210, 242 CrossRef.
- V. Ramesh Babu, K. S. V. Krishna Rao, M. Sairam, B. Vijaya Kumar Naidu, K. M. Hosamani and T. M. Aminabhavi, J. Appl. Polym. Sci., 2006, 99, 2671 CrossRef.
- K. S. V. Krishna Rao, M. C. S. Subha, B. Vijaya Kumar Naidu, M. Sairam, N. N. Mallikarjuna and T. M. Aminabhavi, J. Appl. Polym. Sci., 2006, 102, 5708 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17314k |
| This journal is © The Royal Society of Chemistry 2015 |