
Evidence of blue-shifting N–H⋯N hydrogen bonding despite elongation of the N–H bond†
Abstract
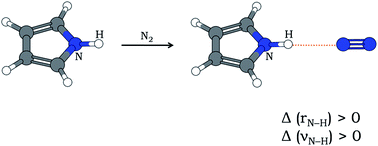
In most cases of X–H⋯Y hydrogen bonding (X is an electronegative atom and Y is any electron-rich center), the X–H bond gets elongated and the stretching frequency of X–H (νX–H) gets red-shifted. Relatively fewer cases of blue-shifting H-bonding are also known, where the X–H bond contracts leading to an increase in νX–H. Here is presented the first experimental report of blue-shifting N–H⋯N H-bonding that accompanies an increase in N–H bond length as evident from quantum chemistry computations. Pyrrole and N2 have been shown to form N–H⋯N hydrogen bond in an ‘annealed’ N2 matrix, where both doped (pyrrole) and dopant (nitrogen) molecules had scope to align themselves and form stable H-bonded clusters. The infrared spectrum of the annealed matrix shows a blue shift of νN–H by 7 cm−1. Computation of anharmonic frequencies at the MP2/6-31+G(d) level of theory reflects the observed blue shift despite N–H bond lengthening after H-bond formation.
 Please wait while we load your content...
Please wait while we load your content...

