
An improved one-pot multicomponent strategy for the preparation of thiazoline, thiazolidinone and thiazolidinol scaffolds†
K. Appalanaidua,
Tulshiram Dadmala,
N. Jagadeesh Babub and
Ravindra M. Kumbhare*a
aFluoroorganic Division, Indian Institute of Chemical Technology, CSIR, Hyderabad, India. E-mail: kumbhare@iict.res.in; Fax: +91-40-27193185
bLaboratory of X-ray Crystallography, Indian Institute of Chemical Technology, CSIR, Hyderabad, India
First published on 2nd October 2015
Abstract
A facile multicomponent synthesis of desirable medicinal scaffolds based on thiazoline, thiazolidinone and thiazolidinol derivatives has been disclosed. The synthesis employs the reaction of a primary amine and carbon disulphide in a microwave, to generate a symmetrical thiourea in situ. The subsequent addition of e.g. 3-bromo-1,1,1-trifluoropropan-2-one affords the desired product, in an efficient one-pot process.
Introduction
Multicomponent reactions (MCRs) are attractive due to their improved efficiency, reduced waste, atom economy, simplicity and rapid access for the synthesis of biologically-active compounds.1 Multicomponent reactions are advantageous over conventional multistep sequences owing to reductions in the costs of reagents, solvents and other resources required for purification and isolation.2 The greater occurrence of infectious diseases and multi-drug-resistant strains has become a major concern in the medicinal area. Therefore, the development of new potential drugs to counteract the increasing resistance is one of the key issues and challenges for medicinal chemistry and related disciplines nowadays. The current scenario highlights the need for the discovery and development of new drugs.In view of the fact that heterocyclic compounds in general, thiazolines, thiazolidinones and thiazolidines exhibit a wide variety of biological activities, the search for new approaches towards the concluding entities with a greater degree of efficiency is of significant importance.
The remarkable physiological activities of thiazolines, thiazolidinones and thiazolidines render them target compounds in organic synthesis. The unsaturated 2-imino-1,3-thiazolines attract great attention in pharmaceutical chemistry due to their important biological activities, such as their antimicrobial, anti-inflammatory, antihistaminic, antihypertensive, hypnotic and anticonvulsant activities, their applicability for the identification of human cells with positive myeloperoxidase reactivity, and their use in agriculture as acaricides, insecticides and plant growth regulators;3,4 they can inhibit melanin production in a dose-dependent manner, thus acting as a skin-whitening agent,5 I (KHG22394, Fig. 1) and act as a reversible inhibitor of p53-mediated apoptosis and p53-dependent gene transcription6 II (Pft-α, Fig. 1). Thiazolidine-4-onethione, a five-membered heterocyclic scaffold, has also gained a lot of attention due to its wide range of applications in medicinal chemistry, such as antidiabetic,7 anticancer,8 calcium-channel blocking9,10 V (Fig. 1), platelet-activating factor (PAF) antagonistic,11 anti-HIV,12 hypnotic,13,14 antitubercular,15 cardiovascular16 III (Fig. 1) and cyclooxygenase (COX) inhibitory activities17 IV (Fig. 1). 2-Imino-1,3-thiazolidines are well recognised for their anti-inflammatory, anodyne and anti-Alzheimer activities, in agriculture as pesticides, and for their protective properties against γ-radiation.18–20
 | ||
| Fig. 1 Some biologically-active scaffolds containing thiazoline and thiazolidinone moieties. | ||
According to the literature, a number of strategies for the synthesis of thiazolines, thiazolidinones and thiazolidines are known. First approaches toward the synthesis of 2-imino-1,3-thiazolines were published more than a century ago, accomplished with condensation reactions of α-haloketones with thiourea, in neutral or basic medium, or with ammonium thiocyanate, also in aqueous media, catalysed by diammonium hydrogen phosphate or DABCO.21 In addition to 2-iminothiazolines, the condensation reactions of α-haloketones with thiourea under acidic conditions gave rise to variable amounts of aminothiazoles as side products.
Murru et al.22 reported the one-pot reaction of 1,1′-(ethane-1,2-diyl)dipyridinium bistribromide (EDPBT), as a brominating agent, enolizable ketones and disubstituted thioureas. Several alternative methods have been developed, which include the use of catalysts such as copper or TiCl4 (ref. 23–30), and other condensation as well as cycloaddition reactions. Recently, one-pot, three-component reactions of aromatic α-bromoketones, primary amines and phenyl isothiocyanate have been reported for the synthesis of thiazol-2-imines using catalytic amounts of triethylamine.31
The general methods for the synthesis of 2-iminothiazolidin-4-ones include the cyclization of thiourea with α-halocarboxylic acids, acyl halides or carboxylic esters, or the condensation reaction of the appropriate amine, aldehyde and mercapto acids.32–39 Recently, thiazolidinone derivatives have been prepared by using CuFe2O4 magnetic nanoparticles as a catalyst.40
Various reports are available for the synthesis of 2-iminothiazolidines, which involves the acid-mediated or, in the presence of triphenylphosphine and diethyl azodicarboxylate, intramolecular cyclization of N-(2-hydroxyethyl)thiourea;41 2-imino-1,3-thiazolidines have also been prepared from treatment of aziridines with thiocyanuric acid, the reaction of 2-vinylaziridine with phenyl isothiocyanate, the condensation of α-halo ketones with thioureas, etc.42–44
As per the literature reviewed, most methods use thiourea or substituted thiourea as a substrate, while in reports of one-pot syntheses, the use of catalysts or base and harsh reaction conditions is required; in aqueous media, low yields have been obtained. To improve on these limitations, it is important to investigate regarding improved strategies for the synthesis of thiazoline, thiazolidinone and thiazolidinol derivatives.
Previously, we successfully used N,N-dimethylformamide (DMF) as a reaction medium and phenylisothiocyanate as a substrate to accomplish the synthesis of 2-imino-4-(trifluoromethyl)thiazolidin-4-ol derivatives.45 As part of our ongoing efforts to explore new routes for heterocyclic synthesis and their biological evaluation,46 herein we report an efficient, one-pot synthesis of thiazolidinol, thiazolidinone and thiazoline derivatives, using carbon disulphide (CS2) as a substrate under microwave irradiation.
Results and discussion
In order to optimize the reaction conditions, we selected the synthesis of (Z)-N-(3,4-diphenylthiazol-2(3H)-ylidene)aniline (4a) as a model reaction (Table 3, entry a). Initially, the reaction of aniline (1 mmol), carbon disulphide (1 mmol) and 2-bromo-1-phenylethanone (1 mmol) was attempted in DMF at room temperature, but there was no progress of the reaction. Later, on conventional heating at 90 °C in DMF for 2 h, we observed a low yield (40%) with some side products. Subsequently, in order to improve the reaction efficiency, we carried out the reaction under microwave irradiation and varied the molar ratios of the reactants; when a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (1:CS2:3, Scheme 1) was used, the product was isolated in the highest yield in a shorter time. The cause for there being no reaction at room temperature, and a lower yield on conventional heating, may be the substantial time required for the formation of thiourea. Most of the literature reports on thiourea notify more than 24 h for thiourea formation. Therefore, we selected the microwave method for the reaction. To further increase the productivity of the reaction, independently, we studied the synthesis of thiourea under various conditions. We attempted the reaction of aniline (2 mmol) and carbon disulphide (1 mmol) under microwave irradiation in various solvent systems (Table 1). Among the various solvents, the reaction proceeded most smoothly in N,N-dimethylformamide (DMF), to give the corresponding thiourea in a 98% yield (Table 1, entry 2). In order to further optimize the reaction conditions for thiourea synthesis, we performed the same reaction in a N,N-dimethylformamide (DMF) solvent under various conditions. Surprisingly, under microwave irradiation, we observed a good yield (98%) of the corresponding thiourea (Table 2, entry 3). Reduction of the time required for the thiourea preparation has constituted the focus of our investigations regarding improved strategies for further synthesis. Microwave irradiation and N,N-dimethylformamide (DMF) thus seemed to be the best choice.
:1:1 (1:CS2:3, Scheme 1) was used, the product was isolated in the highest yield in a shorter time. The cause for there being no reaction at room temperature, and a lower yield on conventional heating, may be the substantial time required for the formation of thiourea. Most of the literature reports on thiourea notify more than 24 h for thiourea formation. Therefore, we selected the microwave method for the reaction. To further increase the productivity of the reaction, independently, we studied the synthesis of thiourea under various conditions. We attempted the reaction of aniline (2 mmol) and carbon disulphide (1 mmol) under microwave irradiation in various solvent systems (Table 1). Among the various solvents, the reaction proceeded most smoothly in N,N-dimethylformamide (DMF), to give the corresponding thiourea in a 98% yield (Table 1, entry 2). In order to further optimize the reaction conditions for thiourea synthesis, we performed the same reaction in a N,N-dimethylformamide (DMF) solvent under various conditions. Surprisingly, under microwave irradiation, we observed a good yield (98%) of the corresponding thiourea (Table 2, entry 3). Reduction of the time required for the thiourea preparation has constituted the focus of our investigations regarding improved strategies for further synthesis. Microwave irradiation and N,N-dimethylformamide (DMF) thus seemed to be the best choice.
 | ||
| Scheme 1 Comparison of previous work with our strategy. | ||


The possible mechanism for thiourea formation is outlined in Scheme 2. The reaction may proceed via the formation of phenylcarbamodithioic acid.
 | ||
| Scheme 2 Plausible mechanism of thiourea formation. | ||
After the optimization, we further studied the effects of the substrate for this method, with different amines subjected to the treatment with CS2 under microwave irradiation followed by addition of various bromo acyl ketones in N,N-dimethylformamide (DMF), and the results are summarized in Table 3. Remarkably, several primary amines, including aniline, benzylamine, furan-2-ylmethanamine, cyclohexylamine, substituted aniline and butylamine, and 2-bromo-1-phenylethanone and 3-(2-bromoacetyl)-2H-chromen-2-one reacted with carbon disulphide, affording the corresponding thiazoline derivatives in the form of HBr salts; these were later neutralized with saturated sodium bicarbonate to achieve the desired thiazoline derivatives (4a–j) in good to excellent yields (Table 3, entries a–j). The plausible reaction mechanism of formation of the products (4a–j) is summarized in Scheme 3. The reaction proceeds via an attack of the thiocarbonyl group of thiourea on the bromomethyl group of the bromoacylketones, followed by cyclization and dehydration processes, affording the products 4a–j.
|
||||
|---|---|---|---|---|
| Entry | R 1 | R1 3 | Product 4b | Yieldc (%) |
| a Reactions were performed with 2 mmol of amine, 1 mmol of CS2 and 1 mmol of phenacyl bromide.b All products were characterized by 1H and 13C NMR, IR and mass spectrometry.c Yield refers to pure product after column chromatography. | ||||
| a |  |
 |
 |
91 |
| b |  |
 |
 |
94 |
| c |  |
 |
 |
88 |
| d |  |
 |
 |
90 |
| e |  |
 |
 |
86 |
| f |  |
 |
 |
88 |
| g |  |
 |
 |
91 |
| h |  |
 |
 |
92 |
| i |  |
 |
 |
90 |
| j |  |
 |
 |
91 |

 | ||
| Scheme 3 Plausible reaction mechanism of thiazoline derivatives. | ||
During the syntheses of the thiazolidinone derivatives, we tried to optimize the reaction conditions. The mixture of a primary amine (2 mmol) and carbon disulphide (1 mmol) was subjected to microwave irradiation for 5 min. After cooling to room temperature, DMF (3 ml) was added to the reaction mixture, and then diethyl but-2-ynedioate or dimethyl but-2-ynedioate (1 mmol) was added. The reaction mixture was stirred at room temperature for 10–15 min. The reaction proceeded smoothly, affording the corresponding thiazolidinone derivatives (6a–i) in good to excellent yield (Table 4, entries a–i). Various amines, including substituted aniline, benzylamine, furfurylamine and butylamine, and diethyl but-2-ynedioate and dimethyl but-2-ynedioate reacted with carbon disulphide, to give the corresponding thiazolidinone derivatives (Table 4). This method was efficiently employed not only for aliphatic and heteroaryl primary amines, but also for aromatic primary amines. The proposed reaction mechanism for formation of the thiazolidinone derivatives is outlined in Scheme 4. The reaction proceeds via the in situ formation of thiourea, followed by the attack of the thiocarbonyl group of thiourea on the acetylene fragment of diethyl but-2-ynedioate, affording the products 6a–i.
|
||||
|---|---|---|---|---|
| Entry | R 1 | R2 2 | Product 6b | Yieldc (%) |
| a The reactions were performed with 2 mmol of amine, 1 mmol of CS2 and 1 mmol of diethyl but-2-ynedioate or dimethyl but-2-ynedioate.b Products were characterized by 1H and 13C NMR, IR and mass spectrometry.c Isolated yield after column chromatography. | ||||
| a |  |
C2H5 |  |
85 |
| b |  |
C2H5 |  |
84 |
| c |  |
C2H5 |  |
90 |
| d |  |
C2H5 |  |
83 |
| e |  |
CH3 |  |
82 |
| f |  |
CH3 |  |
89 |
| g |  |
CH3 |  |
91 |
| h |  |
CH3 |  |
85 |
| i |  |
CH3 |  |
90 |

 | ||
| Scheme 4 Plausible reaction mechanism of thiazolidinone derivatives. | ||
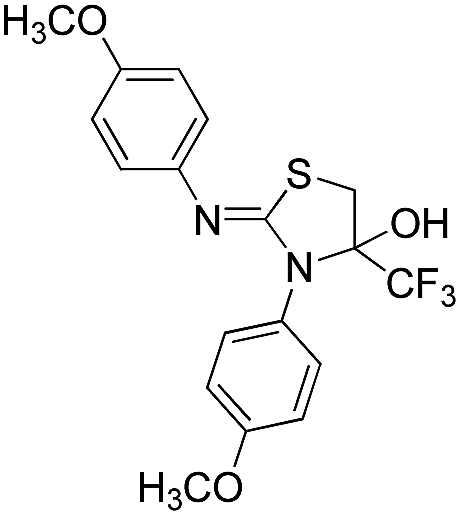
In the case of the syntheses of thiazolidinol derivatives, the reaction of a primary amine and carbon disulphide was performed under optimized microwave conditions, followed by the dropwise addition of 3-bromo-1,1,1-trifluoropropan-2-one in DMF at room temperature, giving the product in good to excellent yield (Table 5, entries 8a–h). The reaction was investigated for various primary amines including benzylamine, furfurylamine, butylamine and substituted anilines. In case of the thiazoline products, after cyclization the tertiary hydroxyl group in the intermediate undergoes a dehydration process. However, during the formation of the thiazolidinol products, the dehydration process does not take place. This might be due to the electron-withdrawing effect of the trifluoromethyl group on the same carbon. The proposed reaction mechanism of formation of the thiazolidinol derivatives is summarized in Scheme 5. The reaction proceeds via thiourea formation, followed by the attack of the thiocarbonyl group of thiourea on the bromomethyl group of 1,1,1-trifluoropropan-2-one, to give products 8a–h.
|
|||
|---|---|---|---|
| Entry | R | Product 8b | Yieldc (%) |
| a Reactions were performed with 2 mmol of amine, 1 mmol CS2 and 1 mmol of 3-bromo-1,1,1-trifluoropropan-2-one.b All products were characterized by 1H and 13C NMR, IR and mass spectrometry.c Yield refers to pure product after column chromatography. | |||
| a |  |
 |
90 |
| b |  |
 |
95 |
| c |  |
 |
91 |
| d |  |
 |
80 |
| e |  |
 |
78 |
| f |  |
 |
95 |
| g |  |
 |
92 |
| h |  |
 |
94 |

 | ||
| Scheme 5 Plausible reaction mechanism of thiazolidinol derivatives. | ||
All the products were characterized by IR spectroscopy, 1H and 13C NMR spectroscopy and mass spectrometry. The structure of 4j (Table 3, entry h) was confirmed by X-ray crystallography, as shown in Fig. 2.47 The X-ray structure shows that compound 4j crystallized in the form of a HBr salt, so we neutralized with saturated sodium bicarbonate to obtain the thiazoline derivatives.
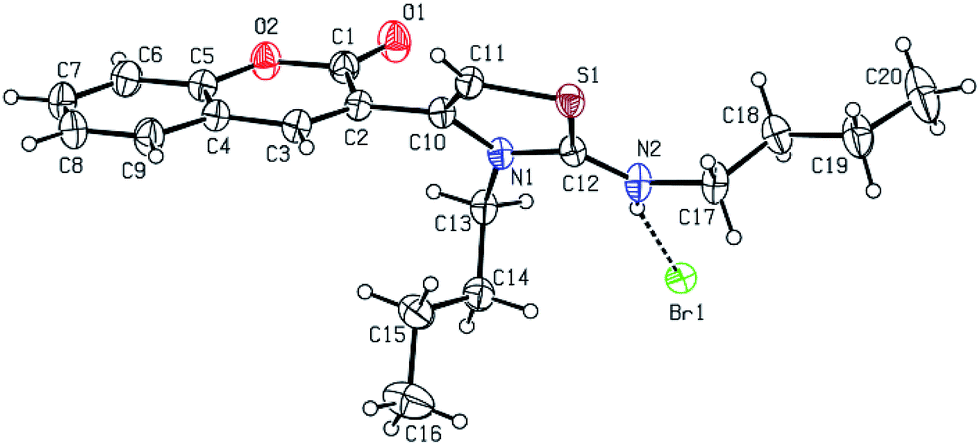 | ||
| Fig. 2 ORTEP molecular diagram of compound 4j. | ||
Conclusions
In summary, we have developed a novel, one-pot and versatile method for the synthesis of thiazoline, thiazolidinone and thiazolidinol derivatives via three-component reactions of a primary amine, carbon disulphide and various bromo acylketones or diethyl but-2-ynedioate or dimethyl but-2-ynedioate, using microwave technology. The reaction uses readily available, inexpensive and harmless precursors. The procedure is extremely useful for synthetic and medicinal chemistry, as it provides the desired products in a single-step process in excellent to good yield.Acknowledgements
We are thankful to the Director of IICT for providing facilities. K. A. N. thank CSIR and T. D. thanks UGC, New Delhi, for the award of a fellowship and R. M. K. thanks S.E.R.C., Department of Science & Technology, Government of India for financial assistance under the Fast Track Scheme for young scientists (SR/FTP/CS-031/2010). We also thanks to Council of Scientific & Industrial Research (CSIR), New Delhi, for financial support under XII five year plan project Fluorochemicals (DITSF) code: CSC-0204.Notes and references
- (a) A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef CAS PubMed; (b) R. V. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958 RSC; (c) C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51 CrossRef CAS; (d) C. Hulme, Multicomponent Reactions, ed. J. Zhu and H. Bienyame, Wiley-VCH, Weinheim, 2005, ISBN 978-3-527-30806-4 Search PubMed.
- (a) A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef CAS; (b) X. Bugaut, T. Constantieux, Y. Coquerel and J. Rodriguez, 1,3-Dicarbonyls in Multicomponent Reactions, in Multicomponent Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M.-X. Wang, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, DOI:10.1002/9783527678174.ch05; (c) Science of Synthesis, Multicomponent Reactions I, ed. T. J. J. Müller, Georg Thieme Verlag KG, Stuttgart, New York, 2014 Search PubMed.
- (a) P. B. Patel and J. T. Trivedi, J. Indian Chem. Soc., 1977, 54, 765 CAS; (b) D. M. Vittoria, O. Mazzoni, E. Piscopo, A. Caligmano and A. Bolognese, J. Med. Chem., 1992, 35, 2910 CrossRef; (c) A. M. Omar and N. H. Eshba, J. Pharm. Sci., 1984, 73, 1166 CrossRef CAS PubMed; (d) M. Chaudhary, S. S. Parmar, S. K. Chaudhari and B. V. Ramasastry, J. Pharm. Sci., 1976, 65, 443 CrossRef CAS PubMed; (e) F. A. Ragab, M. M. Hussain, M. M. Hanna and G. S. Hassan, J. Pharm. Sci., 1993, 82, 387 Search PubMed; (f) H. Y. Hassan, N. A. El-Koussi and Z. S. Farghaly, Chem. Pharm. Bull., 1998, 46, 863 CrossRef CAS; (g) G. Turan-Zitouni, D. M. Sivaci, Z. A. Kaplancikli and A. Ozdemir, Farmaco, 2002, 57, 569 CrossRef CAS; (h) C. G. Bonde and N. J. Gaikwad, Bioorg. Med. Chem., 2004, 12, 2151 CrossRef CAS PubMed; (i) A. Bodtke, W. D. Pfeiffer, N. Ahrens and P. Langer, Bioorg. Med. Chem. Lett., 2004, 14, 1509 CrossRef CAS PubMed.
- (a) F. Nagasaki, T. Yamada, E. Takahashi, Y. Kitagawa and R. Hatano, Jpn. Kokai Tokkyo Koho, JP 63250371, 18/10/1988, Chem. Abstr., 1989, 110, 192810; (b) H. Hoelzel, D. Creuzburg, P. Stohr, H. Dehne, J. Teller, L. Kranz, H. Luthardt, T. Roethling and A. Kaestner, Ger. DD 258168, 13/07/1988, Chem. Abstr., 1989, 111, 2681.
- D. S. Kim, Y. M. Jeong, I. K. Park, H. G. Hahn, H. K. Lee, S. B. Kwon, S. J. Yang, U. D. Sohn and K. C. Park, Biol. Pharm. Bull., 2007, 30, 180 CAS.
- P. Nicolas, M. Anice, D. Rosanna, L. Paul, P. Veronique, L. Fabienne, B. Mathias, K. Jean-Louis and M. Flavio, J. Med. Chem., 2006, 49, 3645 CrossRef PubMed.
- H. Ueno, T. Oe, I. Snehiro and F. Nakamura, US Pat., 5594016, 1997.
- M. Y. Ebeid, O. A. Fathallah, M. I. El-Zaher, M. M. Kamel, W. A. Abdon and M. M. Anwar, Bull. Fac. Pharm., 1996, 34, 125 CAS.
- A. Hara, T. Suzuki, H. Hashizume, N. Shishido, M. Nakamura, F. Ushikubi and Y. Abiko, Eur. J. Pharmacol., 1999, 385, 81 CrossRef CAS.
- T. Kato, T. Ozaki, K. Tamura, Y. Suzuki, M. Akima and N. Ohi, J. Med. Chem., 1999, 42, 3134 CrossRef CAS PubMed.
- Y. Tanabe, G. Suzukamo, Y. Komuro, N. Imanishi, S. Morooka, M. Enomoto, A. Kojima, Y. Sanemitsu and M. Mizutani, Tetrahedron Lett., 1991, 32, 379 CrossRef CAS.
- R. K. Rawal, Y. S. Prabhakar, S. B. Katti and E. de Clercq, Bioorg. Med. Chem., 2005, 13, 6771 CrossRef CAS PubMed.
- S. K. Chaudhari, M. Verma, A. K. Chaturvedi and S. S. Parmar, J. Pharm. Sci., 1975, 64, 614 CrossRef CAS PubMed.
- M. Chaudhary, S. S. Parmar, S. K. Chaudhary, A. K. Chaturvedi and B. V. Rama Sastry, J. Pharm. Sci., 1976, 65, 443 CrossRef CAS PubMed.
- (a) N. M. Turkevich, L. Y. Ladnaya, I. V. Pleshnev and O. M. Grom, Khim, Issled. Farm., 1970, 64, 7 Search PubMed; (b) N. M. Turkevich, L. Y. Ladnaya, I. V. Pleshnev and O. M. Grom, Chem. Abstr., 1972, 76, 34154 Search PubMed.
- S. Nagar, H. H. Singh, J. N. Sinha and S. S. Parmar, J. Med. Chem., 1973, 16, 178 CrossRef CAS.
- R. Ottaná, E. Mazzon, L. Dugo, F. Monforte, R. Maccari, L. Sautebin, G. de Luca, M. G. Vigorita, S. Alcaro, F. Ortuso, A. P. Caputi and S. Cuzzocrea, Eur. J. Pharmacol., 2002, 448, 71 CrossRef.
- (a) M. Takagi, K. Ishimitsu and T. Nishibe, PCT Int. Appl. WO 0172723 A1, 2001, Chem. Abstr., 2001, 135, 288773; (b) B. Latli and J. E. Casida, U.S. 6303638 B1, 16/10/2001, Chem. Abstr., 2001, 135, 303904; (c) M. Tomizawa, A. Cowan and J. E. Casida, Toxicol. Appl. Pharmacol., 2001, 177, 77 CrossRef CAS PubMed.
- (a) A. Elbert, A. Buchholz, U. Ebbinghaus-Kintscher, R. Nauen and H. J. Schnorbach, Pflanzenschutz-Nachrichten Bayer, 2001, 54, 185 (Chem. Abstr., 2002, 136, 274777) CAS; (b) D. A. Schulz-Jander, W. M. Leimkuehler and J. E. Casida, Chem. Res. Toxicol., 2002, 15, 1158 CrossRef CAS PubMed; (c) J. Krohn, Pflanzenschutz-Nachr, Bayer, 2001, 54, 281 (Chem. Abstr., 2002, 136, 228345) CAS; (d) M. Schuld and R. Schmuck, Ecotoxicology, 2000, 9, 197 CrossRef CAS.
- S. J. Hosseinimehr, A. Shaffiee, H. Mozdarani and S. Akhlagpour, J. Radiat. Res., 2001, 42, 401 CrossRef CAS.
- (a) A. Hantzsch and J. H. Weber, Ber. Dtsch. Chem. Ges., 1887, 20, 3118 CrossRef PubMed; (b) V. Traumann, Liebigs Ann. Chem., 1888, 249, 31 CrossRef PubMed; (c) S. Balalaie, S. Nikoo and S. Haddadi, Synth. Commun., 2008, 38, 2521 CrossRef CAS PubMed.
- S. Murru, C. B. Singh, V. Kavala and B. K. Patel, Tetrahedron, 2008, 64, 1931 CrossRef CAS PubMed.
- A. Miloudi, D. El-Abed, G. Boyer, J. Finet, J. Galy and D. Siri, Eur. J. Org. Chem., 2004, 7, 1509 CrossRef PubMed.
- F. Tittelbach, S. Vieth and M. Schneider, Eur. J. Org. Chem., 1998, 1998, 515 CrossRef.
- J. Svetlik, J. Org. Chem., 1990, 55, 4740 CrossRef CAS.
- M. Dhooghe, A. Waterinckx and N. de Kimpe, J. Org. Chem., 2005, 70, 227 CrossRef CAS PubMed.
- Y. Sanemitsu, S. Kawamura, J. Satoh, T. Katayama and S. Hashimoto, J. Pestic. Sci., 2006, 31, 305 CrossRef CAS.
- N. de Kimpe, M. Boelens and J. P. Declereq, Tetrahedron, 1993, 49, 3411 CrossRef CAS.
- S. T. Ingle, S. M. Kapley and M. S. Chande, Proc. - Indian Acad. Sci., Chem. Sci., 1980, 89, 295 CAS.
- (a) X. Wang, F. Wang, Z. Quan, Z. Zhang and M. Wang, Synth. Commun., 2006, 36, 2453 CrossRef CAS PubMed; (b) M. Xia and Y. D. Lu, Synth. Commun., 2006, 36, 1637 CrossRef CAS PubMed; (c) A. Manaka, T. Ishii, K. Takahashi and M. Sato, Tetrahedron Lett., 2005, 46, 419 CrossRef CAS PubMed.
- M. M. Heravi and S. Moghimi, Tetrahedron Lett., 2012, 53, 392 CrossRef CAS PubMed.
- J. Blanchet and J. Zhu, Tetrahedron Lett., 2004, 45, 4449 CrossRef CAS PubMed.
- M. Sedlák, J. Hanusek, V. Macháček and L. J. Hejtmánková, J. Heterocycl. Chem., 2002, 39, 1105 CrossRef PubMed.
- A. Alizadeh, Z. Noaparast, H. Sabahno and N. Zohreh, Helv. Chim. Acta, 2010, 93, 1401 CrossRef CAS PubMed.
- S. Kasmi-Mir, A. Djafri, L. Paquin, J. Hamelin and M. Rahmouni, Molecules, 2006, 11, 597 CrossRef CAS.
- A. Saeed, N. Abbas and U. J. Flörke, J. Braz. Chem. Soc., 2007, 18, 559 CrossRef CAS.
- R. Yella, H. Ghosh and B. K. Patel, Green Chem., 2008, 10, 1307 RSC.
- D. R. St. Laurent, Q. Gao, D. Wu and M. H. Serrano-Wu, Tetrahedron Lett., 2004, 45, 1907 CrossRef CAS PubMed.
- R. K. Rawal, Y. S. Prabhakar, S. B. Katti and E. De Clercq, Bioorg. Med. Chem., 2005, 13, 6771 CrossRef CAS PubMed.
- G. Pal, S. Paul and A. R. Das, New J. Chem., 2014, 38, 2787 RSC.
- (a) A. Hirishima, Y. Yoshii and M. Eto, Agric. Biol. Chem., 1991, 55, 2537 CrossRef; (b) E. Cherbuliez, B. Baehler, O. Espejo, H. Jondra, B. Williahm and J. Rabinovitz, Helv. Chim. Acta, 1967, 50, 331 CrossRef CAS PubMed; (c) T. H. Kim and M. T. Cha, Tetrahedron Lett., 1999, 40, 3125 CrossRef CAS.
- (a) S. Gabriël and J. Colman, Chem. Ber., 1914, 47, 1866 CrossRef PubMed; (b) M. Mousseron, F. Winternitz and R. Dennilauer, Bull. Soc. Chim. Fr., 1956, 1228 Search PubMed; (c) R. A. Wohl and D. H. Headley, J. Org. Chem., 1972, 37, 4401 CrossRef CAS.
- D. C. Butler, G. A. Inman and H. Alper, J. Org. Chem., 2000, 65, 5887 CrossRef CAS PubMed.
- (a) M. M. Campbell and R. C. Craig, J. Chem. Soc., Perkin Trans. 1, 1980, 766, 10.1039/P19800000766; (b) Y. Iwakura, A. Nabeya and T. Nishiguchi, J. Org. Chem., 1967, 32, 2362 CrossRef CAS; (c) T. Nishiguchi, H. Tochio, A. Nabeya and Y. Iwakura, J. Am. Chem. Soc., 1969, 99, 5835 CrossRef; (d) F. A. Vingiello, M. P. Rorer and M. A. Ogliaruso, Chem. Commun., 1971, 329–329, 10.1039/C29710000329; (e) A. Nabeya, T. Shigemoto and Y. Iwakura, J. Org. Chem., 1975, 40, 3536 CrossRef CAS.
- D. Tulshiram, K. Appalanaidu, K. B. Umesh, N. Jagadeesh Babu and R. M. Kumbhare, Eur. J. Org. Chem., 2014, 2014, 2468 Search PubMed.
- (a) K. B. Umesh, D. Tulshiram, K. Appalanaidu, Y. Khageswara Rao, B. N. Jagadeesh and R. M. Kumbhare, Tetrahedron Lett., 2014, 55, 1296 CrossRef PubMed; (b) K. B. Umesh, D. Tulshiram, R. M. Kumbhare and B. Sridhar, J. Heterocycl. Chem., 2014, 51, 1522 CrossRef PubMed.
- The supplementary crystallographic data (CIF file) and the respective ORTEP diagram for this compound have been provided in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1410087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra17278k |
| This journal is © The Royal Society of Chemistry 2015 |