
Palladium complexes with 3-phenylpropylamine ligands: synthesis, structures, theoretical studies and application in the aerobic oxidation of alcohols as heterogeneous catalysts†
Kazem Karami*a,
Nasrin Haghighat Naeinia,
Vaclav Eignerb,
Michal Dusekb,
Janusz Lipkowskic,
Pablo Hervésd and
Hossein Tavakola
aDepartment of Chemistry, Isfahan University of Technology, Isfahan 84156/83111, Iran. E-mail: karami@cc.iut.ac.ir; Fax: +98 3113912350; Tel: +98 3113912351
bInstitute of Physics, Na Slovance 2, 182 21, Praha 8, Czech Republic
cCardinal Stefan Wyszynski University in Warsaw - Faculty of Mathematical and Natural Sciences, Wojcickiego 1/3, 02-093 Warszawa, Poland
dDepartment of Chemistry, University of Vigo, 36310, Vigo, Spain
First published on 12th November 2015
Abstract
The reaction of 3-phenylpropylamine with Pd(OAc)2 by heating in toluene resulted in the nearly square-planar complex trans-[Pd(C6H5(CH2)3NH2)2(OAc)2] (1). Complex 1 reacted with NaCl in methanol to obtain the corresponding product trans-[Pd(C6H5(CH2)3NH2)2Cl2] (2). Treatment of 2 with triphenylphosphine in dichloromethane afforded trans-[Pd(C6H5(CH2)3NH2)2(PPh3)2]2Cl− (3). All the palladium(II) complexes (1–3) were fully characterized by IR and NMR spectroscopy. In addition, the crystal structures of 1 and 2 were determined by single-crystal X-ray diffraction analysis. In these structures, the acetate and chloride ligands are in trans geometry. Density functional theory (DFT) calculations gave bond lengths and angles that were noted as experimental values. Palladium nanoparticles that were derived from complexes (1–3) were supported on cucurbit[6]uril (CB[6]) and identified by X-ray powder diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), inductively coupled plasma analysis (ICP) and high-resolution X-ray powder spectroscopy (HR-XPS). CB[6]-supported palladium nanoparticles (NPs) were used as heterogeneous catalysts for the aerobic oxidation of alcohols to the corresponding aldehydes or ketones without over-oxidation. CB[6]-Pd NPs (3) (prepared from complex 3) show better catalytic activity than CB[6]-Pd NPs (1), (2), as a higher yield was observed with them in a relatively short time. Factors such as the amount of catalyst, solvent, temperature and reaction time were all systematically investigated to determine their effects on the yield of catalytic alcohol oxidation reactions. This catalytic system displayed high activity and selectivity toward alcohols in mild conditions. The catalyst was reused five times without any significant loss of catalytic activity.
Introduction
The selective oxidation of alcohols to the corresponding carbonyl compounds is an important transformation in the chemical industry.1,2 Traditional oxidizing reagents, which have usually required stoichiometric amounts of inorganic oxidants, such as chromate or permanganate, are expensive and have serious toxicity issues associated with them and are not acceptable from the perspective of green chemistry.3,4 Homogeneous transition metal-based catalysts such as palladium(II) complexes are known that can promote this reaction without the use of toxic oxidants.5Hydrogen peroxide is considered as one of the least toxic oxidants, because it gives water as the only by-product. It also conforms to environmental concerns, wherein the development of catalytic systems that use molecular oxygen as the oxidant is of considerable interest.6–8 Molecular oxygen is an inexpensive and abundant oxidant for the oxidation of alcohols.
The most important organic reactions have been performed with homogeneous palladium catalysts. These catalysts possess many merits, such as a high turnover number, high activity and selectivity and excellent yields, but from economic and environmental standpoints, heterogeneous catalysts are highly desirable.9–14 For this purpose, researchers have immobilized palladium complexes and nanoparticles on various supports, such as polymers,15 silica,16 SBA-15/16,17 MCM-41,18 alumina,19 zeolite,20 carbon,21 clay,22 PEG23 and TiO2,24 to create heterogeneous catalysts, because heterogeneous systems are easy to handle and recover.25–33
CB[6] compounds are a family of macrocyclic molecules that can act as both supports and capping agents (Fig. 1). CB[6] have been synthesized by the condensation of formaldehyde with glycoluril34–38 and have two identical portals. At each entrance to the cavity, six polar carbonyl groups are located and provide two negative fringes for bonding to the surface of metals or nanomaterials. In spite of the polar fringed portals, CB[6] is non-polar and therefore nearly insoluble in all common organic solvents and water. In addition, CB[6] is a rigid cyclic structure that has high thermal and chemical stability against many oxidizing agents.39
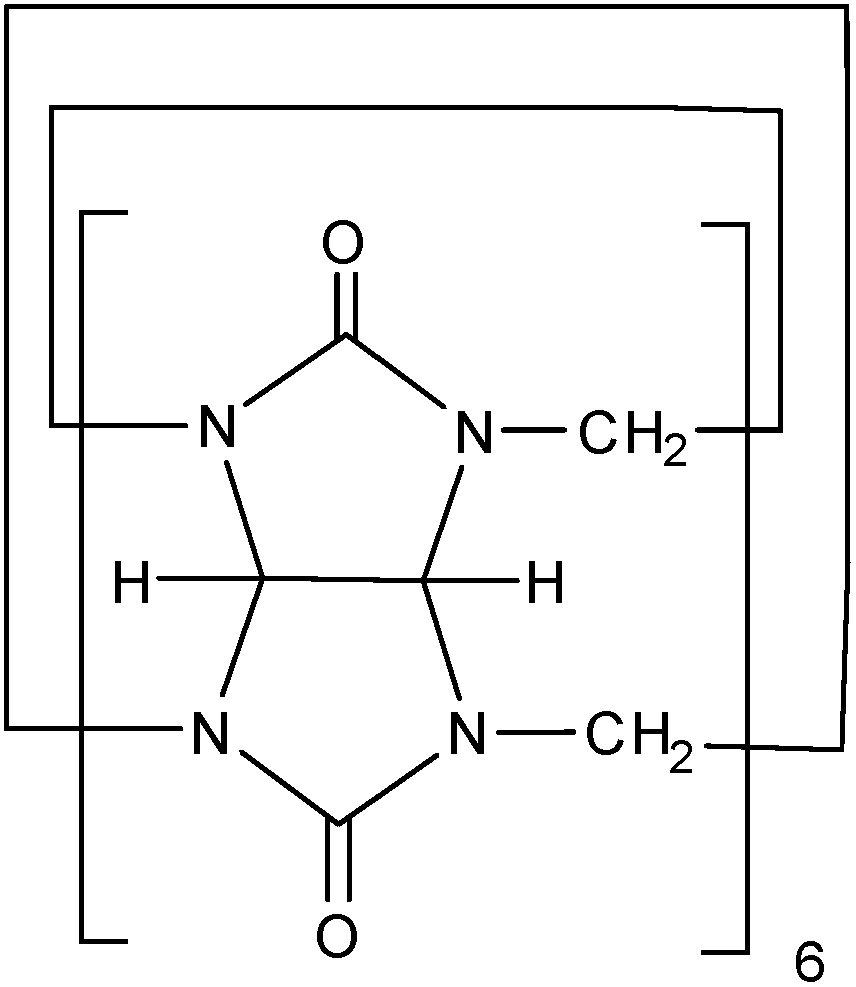 | ||
| Fig. 1 Schematic of cucurbit[6]uril. | ||
Herein, we report the synthesis and characterization of palladium complexes derived from 3-phenylpropylamine. Theoretical studies were also carried out to investigate why we obtained complex 2 instead of orthopalladate (as we expected). The optimized molecular parameters were close to those of the real structure as obtained by X-ray analysis. In addition, the LUMO–HOMO energy gap and partial atomic charges of complex 2 (the real product) and complex 2′ (the desired product) were obtained.
These complexes (1, 2 and 3) were used for the preparation of heterogeneous Pd catalysts, which were deposited on CB[6] using a simple method. The supported Pd nanoparticles showed high activity and selectivity in the aerobic oxidation of alcohols under mild conditions. Finally, the catalyst was fully reusable at least five times.
Results and discussion
Scheme 1 shows the complexes prepared in this study and the labelling assigned to the methylene protons. | ||
| Scheme 1 (i) Pd(OAc)2 in toluene (60 °C, 24 h); (ii) excess NaCl in methanol (room temperature, 12 h); (iii) 2PPh3 in dichloromethane (room temperature, 6 h). | ||
Synthesis and structure of palladium complexes
The reaction of 3-phenylpropylamine Ph(CH2)3NH2 with Pd(OAc)2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio was carried out in toluene at 60 °C to produce the palladium complex 1 trans-[Pd(C6H5(CH2)3NH2)2(OAc)2]. Treatment of complex 1 with an excess of NaCl in methanol afforded the corresponding product trans-[Pd(C6H5(CH2)3NH2)2Cl2] (2). Complex 2 reacted with PPh3 in a 1:2 molar ratio to obtain complex 3 trans-[Pd(C6H5(CH2)3NH2)2(PPh3)2]2Cl−.40 The mononuclear palladium complexes 1, 2 and 3 were characterized by IR and NMR spectroscopy. The spectra for complexes 1, 2 and 3 are given in the ESI (Fig. S1–S9†).
:1 molar ratio was carried out in toluene at 60 °C to produce the palladium complex 1 trans-[Pd(C6H5(CH2)3NH2)2(OAc)2]. Treatment of complex 1 with an excess of NaCl in methanol afforded the corresponding product trans-[Pd(C6H5(CH2)3NH2)2Cl2] (2). Complex 2 reacted with PPh3 in a 1:2 molar ratio to obtain complex 3 trans-[Pd(C6H5(CH2)3NH2)2(PPh3)2]2Cl−.40 The mononuclear palladium complexes 1, 2 and 3 were characterized by IR and NMR spectroscopy. The spectra for complexes 1, 2 and 3 are given in the ESI (Fig. S1–S9†).
The IR spectrum of complex 1 shows characteristic bands that are due to the acetato ligands at 1573 and 1388 cm−1 (ref. 41) and two peaks that correspond to NH2 at 3218 and 3118 cm−1. 1H NMR, 13C{1H} NMR and 31P{1H} NMR spectra of complexes 1, 2 and 3 were recorded in CDCl3. The 1H NMR spectrum of complex 1 shows five sets of signals for methylene protons, acetato-methyl and amine protons. The 13C{1H} NMR spectrum of complex 1 exhibits aliphatic and aromatic regions and the carbon signals of CO and CH3 appear at 180.18 and 23.51 ppm, respectively.
The IR spectrum of complex 2 shows two peaks that correspond to NH2 at 3223 and 3270 cm−1, whereas peaks due to acetato ligands are not present. The 1H NMR spectrum of complex 2 displays four sets of signals for methylene protons and amine protons. The 13C{1H} NMR spectrum of complex 2 shows aliphatic and aromatic regions and the carbon signals of CO and CH3 have disappeared.
The IR spectrum of complex 3 shows two peaks that correspond to NH2 at 3395 and 3451 cm−1. The 1H NMR spectrum of complex 3 displays four sets of signals for methylene protons and amine protons. The 31P{1H} NMR spectrum of complex 3 exhibits a peak that corresponds to PPh3 at 23.26 ppm. Moreover, this spectrum shows an additional peak with lower intensity at 17.86 ppm, which refers to PPh3 in Pd(PPh3)4.42 The molar conductance value of complex 3 in acetone (1 × 10−3 mol cm−3) is 110 Ω−1 cm2 mol−1, which indicates that it is a 1:2 electrolyte.
The single-crystal structures of 1 and 2 were established by X-ray diffraction. However, attempts to crystallize complex 3 were unsuccessful. The single-crystal X-ray diffraction study of complexes 1 and 2 confirmed the details of the proposed new structures.
Fig. 2 and 3 show ORTEP plots of complexes 1 and 2 with selected bond lengths and angles given in the figure caption. Crystal data and parameters concerning data collection and structure solution and refinement are given in Table S1 of ESI.† Tables S2 and S3 in ESI† contain selected bond lengths and angles for complexes 1 and 2, respectively. H atoms in the figures, as well as in the tables, are omitted for clarity. The palladium metal exhibits a distorted square-planar geometry in both complexes and the two acetates and two chlorides are trans to each other.
 | ||
| Fig. 2 ORTEP diagram of trans-[Pd(C6H5(CH2)3NH2)2(OAc)2] (1) with 50% probability ellipsoids. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Pd(1)–O(1), 2.019(9); Pd(1)–O(3), 2.019(10); Pd(1)–N(1), 2.057(12); Pd(1)–N(2), 2.059(14). Selected bond angles (°): O(1)–Pd(1)–N(1), 85.3(5); O(3)–Pd(1)–N(1), 94.3(5); O(3)–Pd(1)–N(2), 85.0(5); O(1)–Pd(1)–N(2), 95.4(5); O(1)–Pd(1)–O(3), 179.4(5); N(1)–Pd(1)–N(2), 179.3(8). | ||
 | ||
| Fig. 3 ORTEP diagram of trans-[Pd(C6H5(CH2)3NH2)2Cl2] (2) with 50% probability ellipsoids. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Pd(1)–N(1), 2.0361(15); Pd(1)–N(1i), 2.0361(15); Pd(1)–Cl(1), 2.3031(5); Pd(1)–Cl(1i), 2.3031(5). Selected bond angles (°): N(1)–Pd(1)–Cl(1), 88.12(4); N(1)–Pd(1)–Cl(1i), 91.88(4); N(1i)–Pd(1)–Cl(1i), 88.12(4); N(1i)–Pd(1)–Cl(1), 91.88(4); Cl(1)–Pd(1)–Cl(1i), 180.0(5); N(1)–Pd(1)–N(1i), 180.0(5). | ||
As indicated by the ligands at the Pd(II) center of complex 1, the angles subtended vary from 85.0(5)° to 95.4(5)° and from 179.3(8)° to 179.4(5)°. The sum of the bond angles around the palladium is 360.0°. The Pd–N bond lengths, 2.057(12) and 2.059(14) Å, in 1 are close to each other and greater than the values, 2.008(7) and 2.021(6) Å, that were reported for palladium complex 2c.43 Moreover, the Pd–O bond distances (2.019(9) and 2.019(10) Å) in 1 were found to be somewhat longer than the values, 1.953(10) and 1.985(6) Å, that were reported for palladium complex 2c.43
As indicated by the ligands at the Pd(II) center of complex 2, the angles are 88.12(4)°, 91.88(4)°, 88.12(4)° and 91.88(4)° for N(1)–Pd(1)–Cl(1), N(1)–Pd(1)–Cl(1i), N(1i)–Pd(1)–Cl(1i) and N(1i)–Pd(1)–Cl(1), respectively. The sum of the bond angles around the palladium is 360.0°. In complex 2, the Pd1–N1 (2.0361(15) Å) bond length is the same as the Pd1–N1i (2.0361(15) Å) distance. The Pd–Cl bond lengths in 2 (2.3031(5) Å) are in the normal range and consistent with the values reported for palladium(II) complexes.44
N–H⋯O interactions in the crystal of 1 result in the formation of a three-dimensional structure, as shown in Fig. 4, whereas the N–H⋯Cl interactions that are shown in Fig. 5 result in the formation of a three-dimensional network in the case of 2.
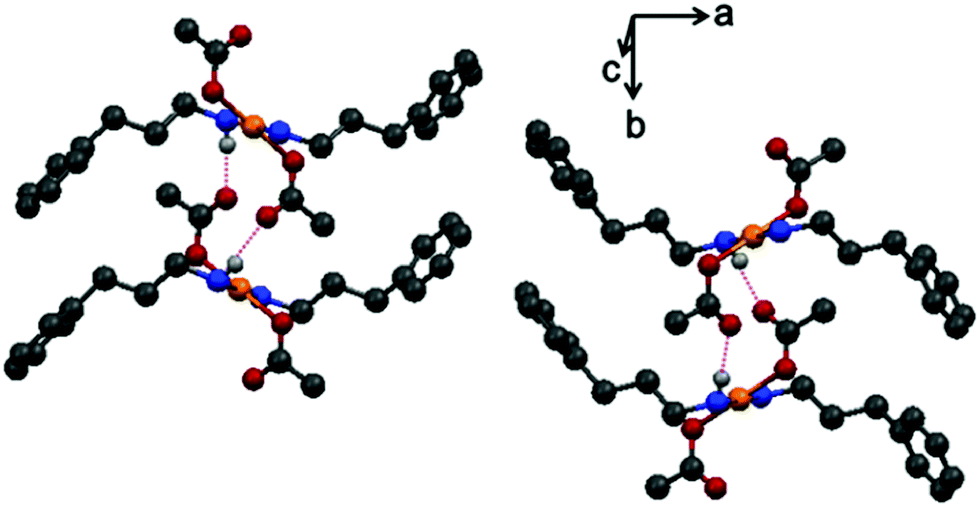 | ||
| Fig. 4 Intermolecular N–H⋯O interactions in complex 1. | ||
 | ||
| Fig. 5 Intermolecular N–H⋯Cl interactions in complex 2. | ||
Synthesis and characterization of CB[6]-Pd NPs
Our goal for the synthesis of CB[6]-Pd NPs was to use simple methods. To achieve this goal, CB[6] was prepared in water and Pd nanoparticles were dispersed on CB[6] in green solvents such as water and ethanol (Scheme 2). In these reactions, palladium complexes 1–3 were used as precursors for preparation of the catalysts.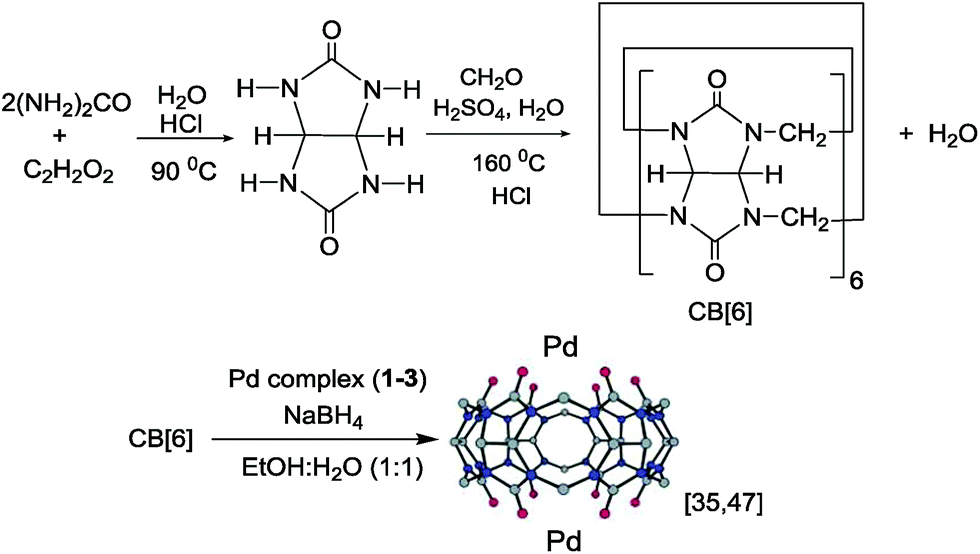 | ||
| Scheme 2 Preparation of CB[6]-Pd NPs 1–3.47 | ||
Previous studies show that CB[6] has two identical portals lined by carbonyl groups, which provide two negative fringes that are capable of binding to the surface of metals or other nanostructures.45,46 Palladium ions can bind to carbonyl groups at the portals of CB[6] before a reducing agent (NaBH4) is added to the reaction mixture.45,46 When a reducing agent is added to the reaction mixture, a high percentage of Pd(II) is transformed into Pd(0). This synthesis procedure is based on the physical adsorption of Pd(0) on the surface of the CB[6] support via electronic interaction between electrons from Pd(0) and vacant orbitals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. However, electrostatic interaction between surface atoms of Pd and CB[6] can effectively prevent the Pd NPs from agglomeration.
O. However, electrostatic interaction between surface atoms of Pd and CB[6] can effectively prevent the Pd NPs from agglomeration.
The XRD pattern of fresh CB[6]-Pd NPs (3) exhibits a broad reflection peak that corresponds to CB[6] at about 23° (ref. 48) and also shows the peaks at 40°, 46°, 68° and 82° that corresponds to (111), (200), (220) and (311) crystallographic planes of Pd(0) nanoparticles with a face-centred cubic Pd lattice (Fig. 6a). This is in good agreement with results obtained from the literature (Fig. 6b).49
 | ||
| Fig. 6 X-ray powder diffraction patterns of (a) CB[6]-Pd NPs (3) and (b) Pd(0).46 | ||
Fig. 7 shows HR-XPS spectra of CB[6]-Pd NPs (3). The Pd 3d spectra show the characteristic Pd 3d5/2 and 3d3/2 doublet peaks. The expected positions of Pd(0) and Pd(II) species, at around 335.3 (ref. 50 and 52) and 337.6 eV,50,51 respectively, are shown.
 | ||
| Fig. 7 XPS spectra of (a) high resolution Pd 3d of fresh CB[6]-Pd NPs (3) and (b) CB[6]-Pd NPs (3) after the fifth run. | ||
The Pd(II) peaks were fitted with a Gaussian–Lorentzian peak shape, whereas the Pd(0) peaks were fitted with an asymmetric shape. The fitting indicates that Pd in fresh CB[6]-Pd NPs (3) is a mixture of Pd(II) and Pd(0) states (Fig. 7a). After the fifth run, Pd(0) is observed in higher proportions, which indicates that after catalytic cycles Pd(II) cations were transformed into Pd(0).
HR-XPS N 1s spectra of fresh CB[6]-Pd NPs (3) and CB[6]-Pd NPs (3) after the fifth run show a single peak at around 400.0 eV, which is attributed to C–N bonds49 from CB[6] and the ligand (Fig. S10†). After the fifth run, C–N bonds are observed in lower proportions, which indicate that after catalytic cycles Pd(II) was transformed into Pd(0) and the ligand was removed from complex 3.
Fig. 8 shows FTIR spectra of palladium complex 3 (a), CB[6] (b) and CB[6]-Pd NPs (c). From Fig. 8a, it is clear that the two bands at around 2924 and 2849 cm−1 could be attributed to asymmetric and symmetric stretching of CH2 groups of 3-phenylpropylamine, respectively. The peak at 3048 cm−1 corresponds to stretching vibrations of CH in the aromatic rings in palladium complex 3. The three strong bands that were observed near 744, 693 and 518 cm−1 in the complex were attributed to the coordinated PPh3 ligand.53 The FTIR spectrum of CB[6] (Fig. 8b) displays a peak at 1726 cm−1. This peak was assigned to carbonyl stretching vibrations, regarding which no changes between CB[6]-Pd NPs (3) (Fig. 8c) and CB[6] (Fig. 8b) were observed, which proves that there is no direct bonding between CB[6] and metal atoms.54 In addition, the absorption bands at 693 and 518 cm−1 demonstrated the presence of PPh3 in CB[6]-Pd NPs (3) in lower proportions (Fig. 8c) compared with palladium complex 3 (Fig. 8a).
 | ||
| Fig. 8 FTIR spectra of: (a) palladium complex 3; (b) CB[6]; (c) CB[6]-Pd NPs. | ||
A FE-SEM image of the corresponding CB[6]-Pd NPs (3) confirms the uniform distribution of Pd NPs on CB[6] (Fig. 9).
 | ||
| Fig. 9 FE-SEM image corresponding to CB[6]-Pd NPs (3). | ||
The corresponding TEM images reveal that the Pd NPs were formed and well dispersed on the surface of the support (Fig. 10). The average particle size of Pd(0) in fresh CB[6]-Pd NPs, based on the sizes of more than 243 particles, was 3.11 nm (Fig. 10a). TEM images after the fifth run are shown in Fig. 10b. The average particle size of Pd(0) in CB[6]-Pd NPs after use was 2.62 nm. Moreover, the TEM images show that aggregation of Pd NPs was not observed even after fifth run.
 | ||
| Fig. 10 TEM images of (a) fresh CB[6]-Pd NPs (3) and (b) CB[6]-Pd NPs (3) after the fifth run: the histograms illustrate the size distribution of Pd nanoparticles. | ||
Aerobic oxidation of alcohols by CB[6]-Pd NPs
The selective oxidation of alcohols to the corresponding carbonyl compounds is one of the most important transformations in the synthesis of fine chemicals. Several excellent catalysts for the oxidation of alcohols to the corresponding aldehydes or ketones have been disclosed, but most of these require heating and toxic oxidants, which violate the requirements of “green chemistry”.We first examined the ability of CB[6]-Pd NPs (Cat 1–3) to catalyze the aerobic oxidation of benzyl alcohol. Initially, we focused our attention on the aerobic oxidation of benzyl alcohol under identical reaction conditions (90 °C, toluene as solvent, K2CO3 as base, in air) to compare catalyst 3 with catalysts 1 and 2 (Table 1).
Although all catalysts were able to produce the corresponding benzaldehyde in good yield within the indicated time, catalyst 3 exhibited higher catalytic activity than that of catalysts 1 and 2 (Table 1, entries 1, 2 and 3). This is due to the fact that by changing the ligands on the transition metal atom, catalytic properties can be altered. Moreover, bulky electron-rich phosphines tend to improve the reaction performance and they also act as catalysts themselves.55–57 For further investigation, a blank experiment with only PPh3 was performed and it was found that PPh3 could not act as a catalyst on its own (Table 1, entry 5). In addition, two different amounts of CB[6]-Pd NPs (3) (0.025 and 0.05 mol% Pd) were used, keeping all other reaction parameters fixed, namely, temperature (90 °C), toluene (6 mmol), K2CO3, and reaction time (23 h) in air. In fact, the amount of catalyst had a significant effect on the oxidation of benzyl alcohol (Table 1, entries 3 and 4). The results are shown in Table 1 and indicate 52% and 60% conversion corresponding to 0.025 and 0.05 mol% Pd catalyst, respectively. The lower conversion of benzyl alcohol to benzaldehyde with 0.025 mol% Pd catalyst may be due to there being fewer catalytic sites. Therefore, 0.05 mol% Pd catalyst was taken to be optimal.
Our further investigation concerned the oxidation of benzyl alcohol using CB[6]-Pd NPs (3), toluene and atmospheric air as a source of molecular oxygen in the presence of various bases (Table 2).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Pd (mol%) | Base | Temp. (°C) | Time (h) | Conv.b (%) |
| a Reaction conditions: benzyl alcohol (1 mmol), base (1 mmol), solvent (6 mL), catalyst (0.025 or 0.05 mol% Pd), in air.b Determined by GC using biphenyl as an internal standard.c Under air bubbling.d Conversion to benzoic acid.e The reaction was carried out in the presence of CB[6].f The reaction was carried out in the presence of Pd(OAc)2 supported on CB[6].g The reaction was carried out in the presence of PdCl2 supported on CB[6]. | ||||||
| 1 | Toluene | 0.05 | K2CO3 | 40 | 4 | 85c |
| 2 | Toluene | — | K2CO3 | 90 | 23 | 10 |
| 3 | Toluene | 0.05 | — | rt | 10 | 75c |
| 4 | Toluene | 0.05 | K2CO3 | rt | 6 | 68c |
| 5 | Toluene | 0.025 | K2CO3 | 90 | 6 | 70c |
| 6 | Toluene | 0.05 | K2CO3 | 90 | 6 | 77c (18)d |
| 7 | Toluene | 0.05 | K2CO3 | 90 | 23 | 60 |
| 8 | Toluene | 0.05 | K3PO4·3H2O | 90 | 23 | 42 |
| 9 | Toluene | 0.05 | NaOH | 90 | 23 | 41 |
| 10 | Toluene | 0.05 | KOH | 90 | 23 | 13 |
| 11 | Toluene | 0.05 | Na2CO3 | 90 | 23 | 28 |
| 12 | EtOH:H2O (4:1) |
0.05 | K2CO3 | 90 | 23 | 31 |
| 13 | EtOH:H2O (2:1) |
0.05 | K2CO3 | 90 | 23 | 43 |
| 14 | EtOH:H2O (1:1) |
0.05 | K2CO3 | 90 | 23 | 55 |
| 15 | Acetone | 0.05 | K2CO3 | 90 | 23 | 4 |
| 16 | DMF | 0.05 | K2CO3 | 90 | 23 | 8 |
| 17 | MeOH | 0.05 | K2CO3 | 90 | 23 | 8 |
| 18 | EtOH | 0.05 | K2CO3 | 90 | 23 | 4 |
| 19 | CH3CN | 0.05 | K2CO3 | 90 | 23 | 3 |
| 20d | Toluene | 0.05 | K2CO3 | 40 | 4 | —c,e |
| 21e | Toluene | 0.05 | K2CO3 | 40 | 4 | 68c,f |
| 22f | Toluene | 0.05 | K2CO3 | 40 | 4 | 54c,g |

It is reasonable that a base deprotonates the alcohol and starts the catalytic reaction; therefore, the oxidation of alcohols over palladium-based catalysts generally needs a base as a promoter.58 Therefore, different bases were used to promote the oxidation of benzyl alcohol and K2CO3 was found to be the most effective (Table 2, entry 7).
In addition, various solvents were investigated in the presence of K2CO3 and it was found that solvents, such as acetonitrile (CH3CN), N,N-dimethylformamide (DMF), acetone, MeOH and EtOH, were not effective (Table 2, entries 15–19). The conversion of benzyl alcohol was low when H2O:EtOH (1:1, 1:2, and 1:4) was used as the solvent (Table 2, entries 12–14) and toluene was the best choice for this catalytic reaction (Table 2, entry 7). In the presence of air, the reaction time needed to be prolonged to 23 h at 90 °C. To reduce the reaction time, the catalytic oxidation of benzyl alcohol was studied by performing the reaction with benzyl alcohol under air bubbling. To determine the effect of air bubbling on the oxidation reaction of benzyl alcohol, three different temperatures (rt, 40 °C and 90 °C) were investigated. The results show that 68%, 85%, and 77% conversion was achieved, corresponding to rt, 40 °C and 90 °C, (Table 2, entries 1, 4 and 6). At an elevated temperature (90 °C), >95% conversion of benzyl alcohol was found with 77% selectivity for benzaldehyde and 18% selectivity for benzoic acid, which is due to over-oxidation of benzaldehyde to benzoic acid at an elevated temperature (Table 2, entry 6). Moreover, in the presence of CB[6] as the catalyst, the reaction did not occur (Table 2, entry 20).
For further investigation, Pd(OAc)2 and PdCl2 were also supported on CB[6] and in comparison with the present catalyst (CB[6]-Pd NPs (3)), provided lower conversion (Table 2, entries 21 and 22).
The scope of the catalytic system was subsequently extended to the aerobic oxidation of a variety of alcohols under optimized conditions (Table 3). Several aromatic alcohols were employed to study the general application of CB[6]-Pd NPs (3) for the oxidation of alcohols and the results are listed in Table 3, entries 1–8. The selectivity for the corresponding aldehyde or ketone was high for all alcohol oxidation reactions. High efficiency was obtained for the oxidation of 2-phenylethanol, wherein 100% conversion was observed at 2 h (Table 3, entry 2). CB[6]-Pd NPs (3) exhibit higher catalytic activity for substituted aromatic alcohols containing electron-donating groups (e.g. –OCH3) than those containing electron-withdrawing groups (such as –NO2) (Table 3, entries 7 and 8). Moreover, this catalytic system was inactive with linear alcohols (Table 3, entries 9 and 10). The results show that CB[6]-Pd NPs (3) were highly active and selective for the oxidation of alcohols that have at least one benzene ring.
|
|||
|---|---|---|---|
| Entry | Substrate | Time (h) | Conv.b (Yield)c (%) |
| a Reaction conditions: alcohol (1 mmol), K2CO3 (1 mmol), toluene (6 mL), catalyst (0.05 mol% Pd), 40 °C, under air bubbling.b Determined by GC using biphenyl as an internal standard.c Isolated yields. | |||
| 1 | Benzyl alcohol | 4 | 85 (81) |
| 2 | 2-Phenylethanol | 2 | 100 (94) |
| 3 | 1-Phenylethanol | 4 | 98 (91) |
| 4 | 4-Hydroxybenzyl alcohol | 4 | 96 (90) |
| 5 | 2-Chlorobenzyl alcohol | 4 | 77 (75) |
| 6 | 2-Hydroxybenzyl alcohol | 4 | 80 (78) |
| 7 | 4-Nitrobenzyl alcohol | 4 | 84 (81) |
| 8 | 4-Methoxybenzyl alcohol | 4 | 90 (83) |
| 9 | 3-Hexanol | 10 | Trace |
| 10 | 2-Methyl-1-propanol | 10 | Trace |

The catalytic activity of complex 3 was also studied in the aerobic oxidation of a variety of alcohols using the optimized heterogeneous reaction conditions. As presented in Table 4, in these heterogeneous systems, different alcohols react to obtain the corresponding products. In comparison with homogeneous systems, the oxidation of alcohols in the presence of CB[6]-Pd NPs (3) (heterogeneous system) could be considered an efficient and versatile catalytic reaction for the aerobic oxidation of a variety of alcohols.
|
|||
|---|---|---|---|
| Entry | Substrate | Time (h) | Conv.b |
| a Reaction conditions: alcohol (1 mmol), K2CO3 (1 mmol), toluene (6 mL), complex 3 (0.05 mol%), 40 °C, under air bubbling.b Determined by GC using biphenyl as an internal standard. | |||
| 1 | Benzyl alcohol | 4 | 88 |
| 3 | 1-Phenylethanol | 4 | 100 |
| 4 | 4-Hydroxybenzyl alcohol | 4 | 93 |
| 5 | 2-Chlorobenzyl alcohol | 4 | 76 |
| 7 | 4-Nitrobenzyl alcohol | 4 | 88 |
| 8 | 4-Methoxybenzyl alcohol | 4 | 93 |
| 9 | 3-Hexanol | 10 | Trace |

Heterogeneity and recyclability
Further experiments were performed to determine the recyclability of the solid catalyst. Consecutive oxidations of benzyl alcohol under air bubbling were also carried out at 40 °C using toluene as the solvent. The results are summarized in Fig. 11. At a palladium loading of 0.05 mol%, CB[6]-Pd NPs (3) afforded a conversion of 85% under air bubbling after 4 h. After the first reaction cycle, the CB[6]-Pd NPs (3) catalyst was recovered by centrifugation and washed with ethanol and water three times. The catalyst was dried overnight at 100 °C and for the second reaction cycle, fresh benzyl alcohol was added and the other conditions were the same as in the first reaction cycle. A conversion of 87% was achieved after 4 h. From the third to the fifth reaction, no significant change was observed in selectivity and only a slight decrease in conversion was observed, which showed that the catalyst was stable and could be regenerated for repeated use. | ||
| Fig. 11 Recyclability test for catalyst in the aerobic oxidation of alcohols. | ||
ICP-OES analysis showed that the palladium content of fresh CB[6]-Pd NPs (3) was 11.9 wt%. The palladium content after five consecutive reaction cycles was almost the same as that of the fresh catalyst and palladium could not be detected in the filtrate after the reaction. Moreover, a hot filtration test confirmed that the reaction did not continue after the catalyst was removed. These results confirm the heterogeneous character of the catalytically active species, which limits pollution of the environment by the catalyst.
Comparison with other studies
The catalytic performance of the catalyst CB[6]-Pd NPs (3) for the oxidation of alcohols was compared with some previously reported results using different types of support (Table 5).15,59–63 As shown in Table 5, in comparison with other supported palladium catalysts, CB[6]-Pd NPs (3) exhibited comparable yields with a low catalyst loading in a short reaction time. In comparison with previous study, this catalytic system provides a higher turnover frequency (TOF). In addition, the preparation process of the catalyst CB[6]-Pd NPs (3) is very simple and environmentally friendly.| Entry | Catalysta | Reaction conditions solvent/base/temp./time/oxidant | Yieldsb/% | TOFb (h−1) | References |
|---|---|---|---|---|---|
| a Data in parentheses indicate mol% Pd used.b Turnover frequency: mmol aldehyde product/(mmol catalyst × h reaction time). | |||||
| 1 | Pd@SBA-15 (0.4) | Toluene/K2CO3/80 °C/3.5 h/O2 | 83 | 59 | 59 |
| 2 | PdNPs/PS (0.5) | Toluene/K2CO3/85 °C/15 h/— | 98 | 13 | 15 |
| 3 | PdHAP-0 (0.6) | Trifluorotoluene/—/90 °C/1 h/O2 | 99 | 165 | 60 |
| 4 | Pd/MIL-101 (1.5) | Toluene/—/80 °C/1.5 h/O2 | 99 | 44 | 61 |
| 5 | MS-PdG-G (1) | Toluene/K2CO3/110 °C/10 h/O2 | 99 | 10 | 62 |
| 6 | Pd(TOP)/MB-H2O2 | Solvent-free/—/80 °C/24 h/— | 29 | 36 | 63 |
| 7 | CB[6]-Pd NPs (0.05) | Toluene/K2CO3/40 °C/4 h/air bubbling | 81 | 405 | This work |
Theoretical approaches
To determine the structure of the prepared molecule 2 in the gas phase, theoretical calculations were performed. In the previous sections, we described the synthetic methods for the preparation of palladium complexes. According to our previous studies,44b with these reaction conditions, we expected to obtain an orthopalladated complex, but X-ray crystal structure analysis confirmed that the geometry was trans instead. Therefore, optimizations of two structures (the real (2) and expected (2′) products) were performed to find a reason why the trans product is formed.The optimized parameters of 2 are in good agreement with the experimental values determined by X-ray analysis. The most important parameters that were calculated for the optimized structure of 2 are listed in Table 6. A comparison between calculated parameters (2*) and X-ray structure parameters (2) confirms that this structure is close to the real structure.
| 2 | 2* | |
|---|---|---|
| Bond lengths | ||
| Pd1–N1 | 2.0361(15) | 2.058 |
| Pd1–N1i | 2.0361(15) | 2.057 |
| Pd1–Cl1 | 2.3031(5) | 2.466 |
| Pd1–Cl1i | 2.3031(5) | 2.466 |
|
||
| Bond angles | ||
| N1–Pd1–Cl1 | 88.12(4) | 89.9 |
| N1i–Pd1–Cl1 | 91.88(4) | 90.1 |
| N1–Pd1–Cl1i | 91.88(4) | 90.1 |
| N1i–Pd1–Cl1i | 88.12(4) | 89.9 |
| Cl1–Pd1–Cl1i | 180.0(5) | 180.0 |
| N1–Pd1–N1i | 180.0(5) | 180.0 |
According to the X-ray structure, complex 2 has a trans geometry and calculation of the Gibbs free energies of both complexes showed that the trans structure (2) could be obtained more easily than the orthopalladate. The calculated values of ΔG for the reactions that produce 2 and 2′ are −263.72 and −132.35 kcal mol−1, respectively, and the ΔH values are −305.40 and 151.25 kcal mol−1 in the gas phase. Therefore, the Gibbs free energy confirms that the trans compound must be the major product.
We also employed population analyses for 2 and 2′ to determine the energies of the frontier molecular orbitals (FMOs). Graphic representations of the LUMO and HOMO and their related energies for both structures are shown in Fig. 12. The energy gaps of 2 and 2′ are 0.0610 and 0.1647 eV, respectively. By comparing the energies of the frontier orbitals, the LUMO–HOMO energy gap in 2 is less than that in 2′, which shows that this structure is more reactive. For a more exact determination of the partial atomic charges, NBO calculations were used. The results of these calculations for complex 2 show that the partial charges of Cl atoms in complex 2 are about −0.5. Moreover, the charge of Pd in this complex is 0, which demonstrates good interaction.
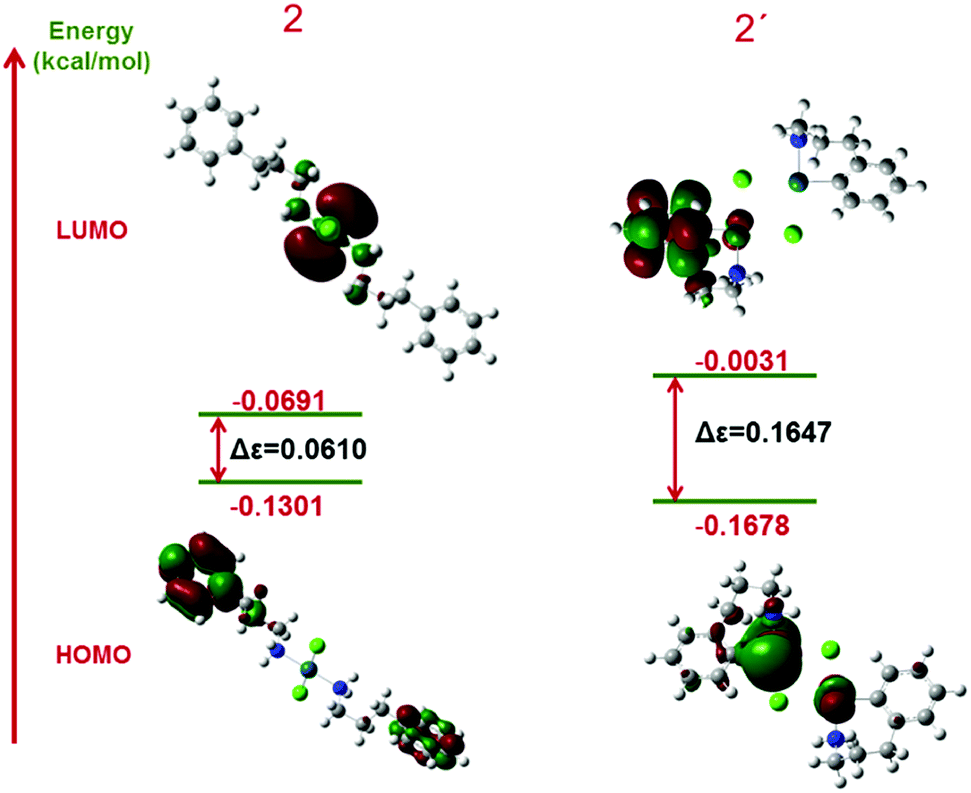 | ||
| Fig. 12 Frontier molecular orbital diagrams of complexes 2 and 2′. | ||
The second-order perturbation energies (E2) that were obtained from the NBO calculations show that the Pd-ligand bonds are stronger in 2 compared with 2′. The E2 interaction energy between Cl and Pd is 172 kcal mol−1 and that between N and Pd is 154 kcal mol−1 for complex 2, whereas the same values for complex 2′ are 142 and 2 kcal mol−1, respectively (Pd–C is a real covalent bond and its energy should be added to the E2 energy). All these calculations confirm the production of complex 2.
Experimental
General
All reactants were purchased from Merck Chemical Company and Aldrich and used as received. Solvents were used without further purification or drying.Fourier transform infrared (FTIR) spectra were obtained in KBr pellets with a Jasco FT/IR 680 Plus instrument. NMR spectra were obtained with a Bruker spectrometer at 400.13 MHz (1H). Elemental analysis was performed on a LECO CHNS-932 apparatus. The molar conductance of complex 3 was measured in acetone at 1 × 10−3 M using an Elmetron CC-505 conductivity meter.
Scanning electron microscopy (SEM) studies were carried out at 15 kV using a HITACHI S-4160 instrument (Japan). Transmission electron micrographs (TEM) were obtained with a JEOL JEM 1010 microscope operating at an accelerating voltage of 100 kV. The palladium content of the catalyst was measured by a inductively coupled plasma (ICP-OES) analyzer (Perkin Elmer 7300DV spectrometer). The X-ray powder diffraction (XRD) pattern was recorded using a Scintag X-ray diffractometer with a 1.54 Å (Cu Kα) X-ray radiation source. HR-XPS experiments were performed using a SPECS Sage HR 100 spectrometer with a non-monochromatic X-ray source (magnesium Kα line with an energy of 1253.6 eV and an applied power of 250 W), which was placed perpendicular to the analyzer axis and calibrated using the 3d5/2 line of Ag with a full width at half maximum (FWHM) of 1.1 eV. An electron flood gun was used during measurements to neutralize charging effects. The resolution that was selected for the spectra of the different elements used pass energy of 15 eV and 0.15 eV per step. All measurements were made in an ultrahigh vacuum (UHV) chamber at a pressure below 8 × 10−8 mbar. In the fittings, Gaussian–Lorentzian and asymmetric functions were used (after a Shirley background correction).
Conversions were monitored using an Agilent Technologies 6890N gas chromatograph equipped with a flame ionization detector (FID) and an HB-50+ column (length = 30 m, inner diameter = 320 μm, and film thickness = 0.25 μm). Products were identified by comparison with authentic samples.
Crystallography
Crystals that were suitable for the X-ray molecular structure determination of 1 and 2 were obtained by the diffusion method for solutions of 1 and 2 in dichloromethane/n-hexane (1:3 v/v) and chloroform/n-hexane (1:3 v/v), respectively. The X-ray diffraction experiment was performed at 120 K with the use of an Agilent Gemini single-crystal diffractometer (Cu Kα radiation). The structure was solved using SuperFlip software64 and further refined with Jana2006.65 MCE software66 was used for visualization of Fourier maps. The structure was refined by the full-matrix least-squares method on the F-squared value. The atoms of the palladium complex were refined anisotropically. The positions of hydrogen atoms were kept in the expected geometry with Uiso set to 1.2 times the Ueq value of the parent atom. The atoms O1, O2 and O3 were refined isotropically and represented disordered solvent. The centers of these atoms do not represent actual atomic positions.
Computational methods
The DFT method was applied to optimize the structures and calculate molecular and spectral parameters of the compounds in the gas phase. The Gaussian 09 program package67 was employed for optimizing the structures and calculation of molecular properties. To perform DFT calculations, Becke's three-parameter exchange functional68 was used in combination with the Lee–Yang–Parr correlation functional (B3LYP) with the LANL2DZ basis set.69 Molecules were used without any symmetry restriction and C1 symmetry was assumed for all molecules. NBO analyses70 were carried out as implemented in the Gaussian program package using the B3LYP/LANL2DZ level of theory.Synthesis of trans-[Pd(C6H5(CH2)3NH2)2(OAc)2] (1)
Pd(OAc)2 (0.0448 g, 0.2 mmol) was added to a solution of 3-phenylpropylamine (28 μL, 0.2 mmol) in toluene (10 mL) and the resulting mixture was heated under reflux for 24 h. The solvent was then evaporated and the resulting yellow solid residue was dissolved in n-hexane (10 mL) and CH2Cl2 (2 mL). A pale yellow solid immediately precipitated. The mixture was stirred for 2 h at room temperature and the resulting suspension was filtered. The pale yellow solid thus obtained was air-dried to obtain (1).Complex 1: yellow solid, yield: 0.06533 g, 66%; mp 135 °C. IR: νmax/cm−1 1573 and 1388 (CO), 3218 and 3118 (NH2). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm): 1.85 (s, 3H, MeCO2), 2.04 (m, 2H, H2), 2.53 (m, 2H, H1), 2.60 (t, 3JHH = 7.6 Hz, 2H, H3), 3.77 (m, 2H, NH2), 7.17–7.30 (m, 5H, C6H5). 13C{1H} NMR (CDCl3, 25 °C, TMS): δ (ppm): 23.51 (Me), 32.50 (C2), 32.90 (C3), 43.23 (C1), 126.15–140.85 (Caromatic), 180.18 (CO). Anal. calcd for C22H32N2O4Pd: C, 53.39; H, 6.52; N, 5.66. Found: C, 53.26; H, 6.65; N, 5.73.
Synthesis of trans-[Pd(C6H5(CH2)3NH2)2Cl2] (2)
To a suspension of 1, methanol was added excess NaCl and the resulting mixture was stirred for 12 h at room temperature. The solvent was then evaporated and the resulting yellow solid residue was dissolved in n-hexane (10 mL) and CH2Cl2 (2 mL). The mixture was stirred for 2 h at room temperature and the resulting suspension was filtered. The pale yellow solid thus obtained was air-dried to obtain (2).Complex 2: yellow solid, yield: 0.04656 g, 52%; mp 170 °C. IR: νmax/cm−1 3223 and 3270 (NH2). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm): 2.03 (m, 2H, H2), 2.67 (m, 2H, H1), 2.69 (m, 2H, H3), 2.78 (m, 2H, NH2), 7.17–7.31 (m, 5H, C6H5). 13C{1H} NMR (CDCl3, 25 °C, TMS): δ (ppm): 32.85 (C2), 33.10 (C3), 44.87 (C1), 126.22–140.65 (Caromatic). Anal. calcd for C18H26Cl2N2Pd: C, 48.29; H, 5.85; N, 6.26. Found: C, 47.79; H, 5.92; N, 6.16.
Synthesis of trans-[Pd(C6H5(CH2)3NH2)2(PPh3)2]2Cl− (3)
To a suspension of palladium complex 2 (0.275 g, 0.5 mmol), dichloromethane (15 mL) was added PPh3 (0.262 g, 1 mmol). The resulting solution was stirred for 6 h and then filtered through a plug of MgSO4. The filtrate was concentrated to ca. 2 mL and n-hexane (10 mL) was added to precipitate 3 as a pale yellow solid, which was collected and air-dried.Complex 3: yellow solid, yield: 0.05047 g, 56%; mp 171 °C (dec.). ΔM (Ω−1 cm2 mol−1)/110. IR: νmax/cm−1 3395 and 3451 (NH2). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm): 1.28 (m, 2H, H2), 2.09 (m, 2H, H1), 2.75 (t, 3JHH = 7.2 Hz, 2H, H3), 3.06 (m, 2H, NH2), 7.28–7.75 (m, C6H5). 31P{1H} NMR (CDCl3, 25 °C): δ (ppm): 23.26 (s, 1P, PPh3 (complex 3)), 17.85 (s, 1P, PPh3 (Pd(PPh3)4)).
Preparation of glycoluril and CB[6]
Glycoluril and CB[6] were prepared according to previous reports.71,72 For the preparation of glycoluril, to a solution of urea (60 g, 1 mol) in water (100 mL) was added a 40% aqueous solution of glyoxal (50 g, 0.345 mol) and concentrated HCl (8.6 mL). The resulting solution was heated at 90 °C until a heavy precipitate was formed. Then, the reaction mixture was allowed to cool to room temperature and filtered. The precipitate was washed with copious amounts of water (200 mL) followed by acetone to remove residual water and dried under vacuum.For the synthesis of CB[6], a stirred mixture of glycoluril (1.5 g, 10.6 mmol), 37% aqueous formaldehyde solution (2.4 mL), concentrated sulfuric acid (1.43 mL) and water (10 mL) was heated for several hours at 160 °C. The resulting reaction mixture was cooled to room temperature and poured into water (25 mL). A pale yellow precipitate was formed, which was filtered off and dissolved in concentrated hydrochloric acid and then the solution was diluted with water. A precipitate was formed, which was washed several times with water and dried at 130 °C.
Preparation of CB[6]-Pd NPs
CB[6]-Pd NPs catalysts were prepared according to the literature method.73 For this purpose, 0.1 mmol palladium complex (1, 2 or 3) and 0.1 mmol (0.099 g) CB[6] were mixed in 10 mL water at room temperature. The mixture was stirred for 30 min until a uniform brown suspension was formed. After this, freshly prepared NaBH4 in ethanol (1 mmol in 10 mL) was rapidly added to the reaction mixture and reduction occurred instantaneously. The reduction was indicated by a color change from brown to black. The mixture was stirred at room temperature for another 3 h. The final reaction product was separated by centrifugation and washed several times with aqueous ethanol (v/v, 1:1) to remove excess salt. CB[6]-Pd NPs were dried at 70 °C for 10 h.
General procedure for the aerobic oxidation of alcohols
Benzyl alcohol (1 mmol), base (1 mmol), solvent (6 mL) and catalyst (0.05 mol% Pd) were combined in a dry flask and the mixture was stirred at a given temperature in air or under air bubbling for a given time. After the reaction, the liquid was filtered and analyzed by GC to determine the conversion and selectivity.Recycling procedure for CB[6]-Pd NPs
Benzyl alcohol was used to test the recyclability of CB[6]-Pd NPs (3) in the aerobic oxidation of alcohols. After the first run was complete, the CB[6]-Pd NPs (3) catalyst was separated by centrifugation and washed several times with ethanol and water to remove adsorbed organic substrates and salt. The catalyst was dried overnight at 70 °C prior to being reused. Then, the catalyst was used for the second run without additional activation and the same process was repeated for the next run.Conclusions
In conclusion, we prepared three palladium complexes that were derived from primary 3-phenylpropylamine, we developed a simple method for their dispersion in the form of palladium nanoparticles on the surface of CB[6], and we tested the application of these nanoparticles for the aerobic oxidation of alcohols in toluene using air as a source of molecular oxygen. The most promising catalytic effects were exhibited by nanoparticles of complex 3, CB[6]-Pd NPs (3), which were evaluated as an efficient catalyst for the aerobic oxidation of alcohols. Importantly, all reactions were performed at 40 °C and the corresponding products were obtained in good or excellent yields. The small size of the Pd particles explains the remarkably high activity of this particular catalyst. TEM and ICP of the regenerated catalyst showed that it is stable under the present reaction conditions.Acknowledgements
This study was supported by the Department of Chemistry, Isfahan University of Technology and the University of Vigo. We gratefully acknowledge the funding support received for this project from the Iranian Nanotechnology Initiative Council. Crystallography was supported by the Czech Science Foundation project 15-12653S.Notes and references
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981, ch. 7, p. 204 Search PubMed.
- M. Hudlicky, Oxidations in Organic Chemistry, ACS Monograph 186, American Chemical Society, Washington, DC, 1990 Search PubMed.
- G. Cainelli and G. Cardillo, Chromium Oxidants in Organic Chemistry, Springer, Berlin, 1984 Search PubMed.
- D. G. Lee and U. A. Spitzer, J. Org. Chem., 1970, 35, 3589 CrossRef CAS.
- M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227 CrossRef CAS.
- D. C. Ebner, J. T. Bagdanoff, E. M. Ferreira, R. M. McFadden, D. D. Caspi, R. M. Trend and B. M. Stoltz, Chem.–Eur. J., 2009, 15, 12978 CrossRef CAS PubMed.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed.
- M. Beller, Chem. Ing. Tech., 2006, 78, 1061 CrossRef CAS.
- N. Selander and K. J. Szabo, Chem. Rev., 2011, 111, 2048 CrossRef CAS PubMed.
- C. E. Tucker and J. G. de Vries, Top. Catal., 2002, 19, 111 CrossRef CAS.
- B. M. Bhanage and M. Arai, Catal. Rev.: Sci. Eng., 2001, 43, 315 CAS.
- S. J. Sabounchei, M. Ahmadi, Z. Nasri, E. Shams and M. Panahimehr, Tetrahedron Lett., 2013, 54, 4656 CrossRef CAS.
- C. Liu, Q. Ni, F. Bao and J. Qiu, Green Chem., 2011, 13, 1260 RSC.
- K. Karami, M. Ghasemi and N. Haghighat Naeini, Catal. Commun., 2013, 38, 10 CrossRef CAS.
- (a) B. Karimi, A. Zamani, S. Abedi and J. H. Clark, Green Chem., 2009, 11, 109 RSC; (b) B. Karimi, A. Zamani and J. H. Clark, Organometallics, 2005, 24, 4695 CrossRef CAS; (c) D. Choudhary, S. Paul, R. Gupta and J. H. Clark, Green Chem., 2006, 8, 479 RSC; (d) C. M. A. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636 CrossRef CAS.
- (a) G. Zhang, Y. Wang, X. Wen, C. Ding and Y. Li, Chem. Commun., 2012, 2979 RSC; (b) H. Yang, X. Han, Z. Ma, R. Wang, J. Liu and X. Ji, Green Chem., 2010, 12, 441 RSC.
- B. Qi, Y. Wang, L. Lou, L. Huang, Y. Yang and S. Liu, J. Mol. Catal. A: Chem., 2013, 370, 95 CrossRef CAS.
- (a) H. Wu, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2005, 347, 1356 CrossRef CAS; (b) C. M. A. Parlett, L. J. Durndell, K. Wilson, D. W. Bruce, N. S. Hondow and A. F. Lee, Catal. Commun., 2014, 44, 40 CrossRef CAS.
- F. Li, Q. Zhang and Y. Wang, Appl. Catal., 2008, 334, 217 CrossRef CAS.
- G. An, H. Ahn, K. A. de Castro and H. Rhee, Synthesis, 2010, 477 CAS.
- T. Hara, J. Sawada, Y. Nakamura, N. Lchikuni and S. Shimazu, Catal. Sci. Technol., 2011, 1, 1376 CAS.
- K. Karami, Z. Karami Moghadam and M. Hosseini-Kharat, Catal. Commun., 2014, 43, 25 CrossRef CAS.
- K. Karami, M. Bahrami Shehni and N. Rahimi, Appl. Organomet. Chem., 2013, 27, 437 CrossRef CAS.
- N. Jamwal, M. Gupta and S. Paul, Green Chem., 2008, 10, 999 RSC.
- M. J. Gronnow, R. Luque, D. J. Macquarrie and J. H. Clark, Green Chem., 2005, 7, 552 RSC.
- N. Erathodiyil, S. Ooi, A. M. Seayad, Y. Han, S. S. Lee and J. Y. Ying, Chem.–Eur. J., 2008, 14, 3118 CrossRef CAS PubMed.
- B. Karimi, S. Abedi, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2006, 45, 4479 CrossRef PubMed.
- Y. Wan, H. Y. Wang, Q. F. Zhao, M. Klingstedt, O. Terasaki and D. Y. Zhao, J. Am. Chem. Soc., 2009, 131, 4541 CrossRef CAS PubMed.
- K. Sarkar, M. Nandi, M. Islam, M. Mubarak and A. Bhaumik, Appl. Catal., A, 2009, 352, 81 CrossRef CAS.
- B. Blanco, M. Brissart, M. Moreno-Mãnas, R. Pleixats, A. Mehdi, C. Reyé, S. Bouquillon, F. Hénin and J. Muzart, Appl. Catal., A, 2006, 297, 117 CrossRef CAS.
- D. Trong On, D. Desplantier-Giscard, C. Danumah and S. Kaliaguine, Appl. Catal., A, 2001, 22, 299 CrossRef.
- P. Taladriz-Blanco, P. Hervés and J. Pérez-Juste, Top. Catal., 2013, 56, 1154 CrossRef CAS.
- R. Behrend, E. Meyer and F. Rusche, Justus Liebigs Ann. Chem., 1905, 1, 339 Search PubMed.
- W. A. Freeman, W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367 CrossRef CAS.
- P. Cintas, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 17, 205 CrossRef CAS.
- A. I. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2002, 66, 8094 CrossRef.
- J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC.
- J. Vicente, A. Arcas, D. Bautista and M. Carmen Ramírez de Arellano, J. Organomet. Chem., 2002, 663, 164 CrossRef CAS.
- H. Onoue and I. Moritani, J. Organomet. Chem., 1972, 43, 431 CrossRef CAS.
- T. Tang, T. Metanawin, A. Hebden, P. McGowan and X. S. Wang, Chem. Commun., 2010, 46, 6663 RSC.
- J. Vicente, I. Saura-Llamas, M. G. Palin, P. G. Jones and M. C. Ramírez de Arellano, Organometallics, 1997, 16, 826 CrossRef CAS.
- (a) F. Saleem, G. K. Rao, A. Kumar, S. Kumar, M. P. Singh and A. K. Singh, RSC Adv., 2014, 4, 56102 RSC; (b) K. Karami, M. Hosseini Kharat, C. Rizzoli and J. Lipkowski, J. Organomet. Chem., 2013, 728, 16 CrossRef CAS.
- H.-J. Buschmann, E. Cleve, L. Mutihac and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 293 CrossRef CAS.
- A. L. Koner, C. Márquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545 CrossRef CAS PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS PubMed.
- Q. Yang, Y. Jiang, X. Li, Y. Yang and L. Hu, Bull. Mater. Sci., 2014, 37, 1167 CrossRef CAS.
- S. Martinez, A. Vallribera, C. L. Cotet, M. Popovici, L. Martin, A. Roig, M. Moreno-Manas and E. Molins, New J. Chem., 2005, 29, 1342 RSC.
- G. Ding, W. Wang, T. Jiang and B. Han, Green Chem., 2013, 15, 3396 RSC.
- J. Yang, D. Wang, W. Liu, X. Zhang, F. Bian and W. Yu, Green Chem., 2013, 15, 3429 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics, Eden Prairie, MN, 1995 Search PubMed.
- S. Harder, M. G. B. Drew and S. Bhattacharya, J. Chem. Sci., 2008, 120, 441 CrossRef.
- M. Cao, J. Ling, H. Yang and R. Cao, Chem. Commun., 2010, 46, 5088 RSC.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366 CrossRef CAS PubMed.
- S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871 CrossRef CAS PubMed.
- K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854 RSC.
- B. Karimi, S. Abedi, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2006, 45, 4776 CrossRef CAS PubMed.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS PubMed.
- G. Chen, S. Wu, H. Liu, H. Jiang and Y. Li, Green Chem., 2013, 15, 230 RSC.
- H. Yang, Z. Ma, Y. Qing, G. Xie, J. Gao, L. Zhang, J. Gao and L. Du, Appl. Catal., A, 2010, 382, 312 CrossRef CAS.
- E. J. García-Suárez, M. Tristany, A. B. García, V. Collière and K. Philippot, Microporous Mesoporous Mater., 2012, 153, 155 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS.
- J. Rohlicek and M. Husak, J. Appl. Crystallogr., 2007, 40, 600 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, J. Izmaylov, A. F. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. K. N. Brothers, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- Modern Supramolecular Chemistry: Strategies for Macrocycle synthesis, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCN, 2008, ch. 4, pp. 137–139 Search PubMed.
- K. Jansen, H. J. Buschmann, A. Wego, D. Dopp, C. Mayer, H. J. Drexler, H. J. Holdt and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 357 CrossRef CAS.
- M. Cao, Y. Wei, S. Gao and R. Cao, Catal. Sci. Technol., 2012, 2, 156 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and FT-IR spectra of all complexes (1–3); single-crystal data of 1 and 2 (CCDC 1060354 and 1060510), HR-XPS N 1s spectra of fresh CB[6]-Pd NPs (3) and CB[6]-Pd NPs (3) after the fifth run. CCDC 1060354 and 1060510. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra17249g |
| This journal is © The Royal Society of Chemistry 2015 |