
Ionic liquid-assisted synthesis of morphology-controlled TiO2 particles with efficient photocatalytic activity†
Baohua Zhanga,
Zhimin Xueb,
Yiguo Xuec,
Zhaodan Huanga,
Zhonghao Li*a and
Jingcheng Haoa
aKey Laboratory of Colloid and Interface Chemistry, Shandong University, Ministry of Education, Jinan 250100, China. E-mail: zhonghaoli@sdu.edu.cn; Fax: +86 531 88564750; Tel: +86 531 88363821
bBeijing Key Laboratory of Lignocellulosic Chemistry, College of Materials Science and Technology, Beijing Forestry University, Beijing 100083, China
cGeotechnical and Structural Engineering Research Center of Shandong University, Jinan 250061, China
First published on 18th September 2015
Abstract
An ionic liquid-assisted synthesis route is proposed for the synthesis of anatase TiO2 particles. Flowerlike TiO2 particles, TiO2 nanosheets and porous TiO2 aggregates can be controllably synthesized by the ionic liquid 1-ethyl-3-methylimidazolium acetate ([EMIM]Ac). The photocatalytic activity towards the degradation of Rhodamine B (RhB) molecules is investigated among the synthesized TiO2 particles in comparison with the commercial Degussa-P25 TiO2. In particular, the as-prepared TiO2 nanosheets exhibit excellent photocatalytic activity, which is higher than the commercial Degussa-P25 TiO2, demonstrating their promising application towards the degradation of organic molecules. Thus, the present work provides a facile route for the synthesis of TiO2 particles with efficient photocatalytic activity.
Introduction
Ionic liquids (ILs) have gained great attention in inorganic nano-object synthesis since the unique adaptability and flexibility of ILs provide materials chemists with a new, flexible, and powerful tool for the fabrication of novel, interesting, and highly sophisticated nanostructures via chemical approaches.1–26 Particularly, inorganic synthesis in ionic liquids occasionally leads to materials that are difficult to achieve using other synthesis routes. For example, unusual materials such as ZnO mesocrystals27 Fe3S4 superstructures28 and metal–organic framework spheres29 have been successfully synthesized based on ionic liquids. In the previous reported researches, ionic liquids can be used as a solvent, as a crystal growth modifier, or as a reactant precursor for the fabrication of inorganics.1,4,30 Up to date, the ionic liquid-assisted synthesis of inorganic particles are mainly limited in ionic liquid–water mixture system.4 Our recent work demonstrates that the ionic liquid–benzyl alcohol system is very promising to synthesize unusual Pd particles with improved electrocatalytic activity31 and complicated WO3 particles32 with enhanced gas sensing properties. Therefore, it is of great interest to explore the ionic liquid–benzyl alcohol system for the synthesis of other inorganic particles with other interesting properties.TiO2 has a wide range of applications such as bioseparation, sensing, energy storage, solar energy conversion, and catalysis.33–37 Therefore, much recent effort has been concentrated on the fabrication of the corresponding nanostructured TiO2 particles to enhance their performance in currently existing applications.38 Until now, there are some reports about the synthesis of nanostructured TiO2 based on ionic liquids.39–47 TiO2 particles such as mesoporous aggregates,39 hollow microspheres,40 uniform nanocolloids,9 nanorods,11 nanoparticles with controllable phase,41,42 fluorinated B/C-codoped TiO2 nanocrystals,43 poly(methyl methacrylate)-TiO2 nanocomposite,44 nitrogen, carbon and fluorine-codoped rutile TiO2 nanorods,45 cellulose-TiO2 nanocomposites46 and anatase TiO2 nanocubes47 have already been synthesized with ionic liquids. The reported TiO2 synthesis is performed in either ionic liquid medium or ionic liquid–water mixture medium. However, there is no report on the synthesis of TiO2 based on the ionic liquid–organic solvent system, which might bring new opportunity for fabrication of interesting TiO2 with improved properties. Specifically, the facile routes to synthesize nanostructured TiO2 particles with controlled morphologies are still open for challenge based on ionic liquids.
Herein, a facile route is proposed for the synthesis of anatase TiO2 particles with controlled morphologies with the assistance of the ionic liquid 1-ethyl-3-methylimidazolium acetate ([EMIM]Ac) in benzyl alcohol solution. Flowerlike TiO2 particles, TiO2 nanosheets and porous TiO2 aggregates are controllably synthesized with the assistance of [EMIM]Ac. In comparison with commercial Degussa-P25 TiO2, the as-prepared TiO2 nanosheets exhibit excellent photocatalytic activity towards the degradation of Rhodamine B (RhB) molecules, demonstrating their promising application towards the degradation of organic molecules. Thus, the present work provides a facile route for the controllable synthesis of anatase TiO2 particles with enhanced photocatalytic activity.
Experimental section
Materials
Titanium isopropoxide and Rhodamine B (RhB) were purchased from Aladdin Industrial Corporation. 1-Ethyl-3-methylimidazolium acetate ([EMIM]Ac) was purchased from Lanzhou Greenchem, ILS, LICP, CAS, China. Degussa-P25 TiO2 was purchased from Alfa Aesar. All the reagents were used without further purification.Synthesis
In a typical synthesis with the mole ratio of titanium isopropoxide to [EMIM]Ac at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:10 and 1:100, firstly 0.07 mmol titanium isopropoxide was dissolved in 8 mL benzyl alcohol. Subsequently, 0.07, 0.7 and 7 mmol [EMIM]Ac was added into the above solution under continuous stirring to result in the homogeneous solution with the [EMIM]Ac concentration at 8.75 mM, 87.5 mM and 875 mM. Then, the resultant solution was put into a Teflon-lined autoclave. After this, the autoclave was heated at 180 °C for 12 h. The autoclave was then cooled to room temperature naturally. Finally, the resultant products were recovered by repeated centrifugation and washing with ethanol and water, respectively, and dried at 60 °C for 5 h. To remove the residual organics from the sample and simultaneously achieve highly crystalline pure TiO2 products, the obtained product was further calcined at 600 °C for 3 h in air to obtain the product. The temperature was increased from room temperature to 600 °C at a heating rate of 2 °C min−1.
:1, 1:10 and 1:100, firstly 0.07 mmol titanium isopropoxide was dissolved in 8 mL benzyl alcohol. Subsequently, 0.07, 0.7 and 7 mmol [EMIM]Ac was added into the above solution under continuous stirring to result in the homogeneous solution with the [EMIM]Ac concentration at 8.75 mM, 87.5 mM and 875 mM. Then, the resultant solution was put into a Teflon-lined autoclave. After this, the autoclave was heated at 180 °C for 12 h. The autoclave was then cooled to room temperature naturally. Finally, the resultant products were recovered by repeated centrifugation and washing with ethanol and water, respectively, and dried at 60 °C for 5 h. To remove the residual organics from the sample and simultaneously achieve highly crystalline pure TiO2 products, the obtained product was further calcined at 600 °C for 3 h in air to obtain the product. The temperature was increased from room temperature to 600 °C at a heating rate of 2 °C min−1.
Characterization
X-ray diffraction (XRD) was performed using a Rigaku Dmax-rc X-ray diffractometer with Ni-filtered Cu Kα (λ = 1.5418 Å) radiation. Transmission electron microscopy (TEM) was performed using a JEM 1400 TEM operating at 120 kV. Scanning electron microscopy (SEM) was performed using a Hitachi SU-70 FESEM operating at 10 kV. FTIR spectra were performed on Nicolet 6700 Fourier transform infrared spectrometer. Thermogravimetric analysis (TGA) was carried out on a SDTA851 thermoanalyzer. N2 adsorption–desorption isotherms were measured on a QuadraSorb SI apparatus. UV-vis spectra were recorded on a U-4100 spectrometer. The X-ray photoelectron spectroscopy (XPS) analysis was carried on an ESCALAB 250 photoelectron spectrometer. Fluorescence spectroscopy measurements were carried out on an LS-55 spectrofluorometer.Photocatalytic tests
The photocatalytic activity of the as-prepared TiO2 particles was evaluated by its photodegradation rate on RhB in deionized water. The catalyst (5 mg) was dispersed in an aqueous RhB solution (25 mL, 1 × 10−5 M) in a reactor cell and the solution was stirred in dark conditions for 1 h to achieve adsorption equilibrium between the dye and the catalyst prior to irradiation. A high pressure Hg lamp (CHF-XM500) was used as the irradiation source and no additional filters were used. After each reaction, the solution was separated from catalyst by centrifugation. The concentration of RhB was measured with a UV-vis spectrophotometer. The concentration of RhB, measured by the intensity of the absorption peak at 553 nm, was followed as a function of time in order to study the kinetics of the reaction.Results and discussion
Fig. 1A and B show XRD patterns of the typical synthesized TiO2 products before and after the calcination at 600 °C. It is found that all of the XRD diffraction peaks can be well indexed to anatase TiO2 (JCPDS card no. 21-1272). However, it is obvious that the particles are more highly crystallized after calcinations compared with initial ones. The XRD results thus confirm the formation of pure anatase TiO2 particles by the present synthesis route. | ||
| Fig. 1 XRD patterns of the TiO2 samples before (A) and after calcinations (B). The TiO2 samples are synthesized with 8.75 mM (a), 87.5 mM (b), 875 mM (c) and 0 mM (d) [EMIM]Ac ionic liquid. | ||
Fig. 2a and b shows the typical low and high magnification SEM images of the initial TiO2 products synthesized with 87.5 mM [EMIM]Ac, manifesting the formation of nanosheet particles with the average size of 113 ± 14 nm. Fig. 2c shows the corresponding SEM image of the as-prepared TiO2 products after calcination at 600 °C. It is found that the TiO2 particles preserve well the nanosheet morphologies although the surfaces of the nanosheets are much rougher than the initial ones. Thus, the calcination process does not obviously influence the morphology of the particles. However, the calcined particles are much more highly crystallized in comparison with the initial particles as discussed in XRD results. The detailed structure of the calcined TiO2 nanosheet particles is shown in the TEM image of Fig. 2d. It is clear that the nanosheets are constructed by the nanoparticle subunits. The size of the nanoparticle subunits is 10 ± 2 nm. Based on the contrast of the TEM image, it is obvious that the nanosheets have a porous structure. In order to understand whether the present [EMIM]Ac ionic liquid plays a unique role for the formation of these nanosheet particles, the controlled synthesis without [EMIM]Ac ionic liquid is performed with the synthesis condition similar to that of nanosheet particles. XRD pattern of the as-prepared TiO2 particles without the assistance of [EMIM]Ac ionic liquid demonstrates the formation of anatase TiO2 (curve d in Fig. 1A). Fig. 2e and f show the SEM and TEM images of the as-prepared particles synthesized without [EMIM]Ac. It is clear that spherical particles with the size of 11 ± 2 nm form. Therefore, it is clear that the ionic liquid has an important role for the formation of the nanosheet particles. To uncover the formation mechanism of these TiO2 nanosheet particles, we recover the products at different reaction time (1 h, 5 h, 8 h, 10 h). Fig. S1a (ESI†) shows XRD patterns of the recovered products at different reaction time. At 1 h, amorphous particles form as demonstrated in the XRD pattern. However, the crystallinity increases with the time prolonged. Beginning with 8 h, typical anatase TiO2 forms as shown in Fig. S1.† Fig. S1(b–e)† show the typical SEM images for the synthesized TiO2 particles at different reaction time. At 1 h, it is found that irregular particles form. At 5 h, nanosheet particles start to form. For the product recovered at 8 h, more nanosheet particles form. Further increasing the time to 10 h, it is found all the products exhibit nanosheet morphologies. In principle, the crystal growth process consists of nucleation and growth, which are influenced by the intrinsic crystal structure and the external conditions including the kinetic energy barrier, temperature, time, capping molecules, and so forth.48 The initial formed nuclei can further growth by consuming the reactants or the metastable primary particles. The growth by consuming metastable primary particles is mainly related to the well-known Ostwald ripening process which is usually apparent in high temperature synthesis. In this process, the [EMIM]Ac could adsorb on the TiO2 crystal to modify the growth preferably to result in the nanosheet morphology. Besides, it is known that 1,3-dialkylimidazolium ionic liquids can form a polymeric supramolecular structure.49,50 Therefore the organized structure of the ionic liquid may also have a template effect, which further helps the formation of the TiO2 nanosheet nanostructures. As a result, the nanosheet nanostructured TiO2 particles could be successfully formed with the assistance of [EMIM]Ac ionic liquid.
 | ||
| Fig. 2 SEM images of the TiO2 sample synthesized with 87.5 mM [EMIM]Ac ionic liquid before (a and b) and after calcination (c). The corresponding TEM image of the calcined sample is shown in (d). SEM (e) and TEM image (f) of the TiO2 sample synthesized without [EMIM]Ac ionic liquid. | ||
In order to verify whether the [EMIM]Ac adsorbs on the surface of the TiO2 for modifying the preferable crystal growth, FTIR characterization is performed on the initial synthesized TiO2 nanosheet products before calcination. For comparison, the FTIR characterization of pure ionic liquid [EMIM]Ac is also performed. Comparing Fig. 3a with Fig. 3b, it is clear that the [EMIM]Ac is doped in the initial nanosheet product. Thus, it demonstrates that the adsorbing of [EMIM]Ac on the TiO2 crystals contributes to the successful formation of the nanosheet structure.
 | ||
| Fig. 3 FTIR spectrum (a) of the TiO2 sample synthesized with 87.5 mM [EMIM]Ac ionic liquid before calcination. FTIR spectrum (b) of the pure [EMIM]Ac ionic liquid. | ||
TGA measurement was further used to determine the weight of organic species adsorbed on the initial TiO2 nanosheet particles before calcination. From Fig. 4, it shows that two weight loss regions appear in the TGA curve. The weight loss of 2.4% below 150 °C is attributed to the evaporation of water in nanosheet particles. Such water comes from the washing process of the products during the recovery procedure. The further weight loss of 15.1% at higher temperature range can be ascribed to the existence of organic species.
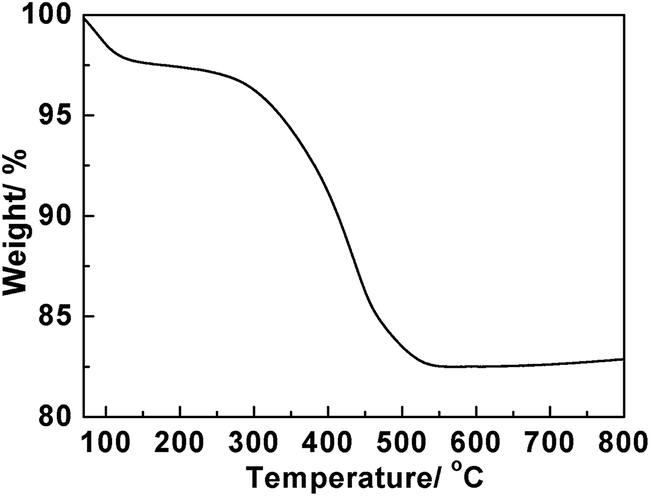 | ||
| Fig. 4 TGA curve of the TiO2 sample synthesized with 87.5 mM [EMIM]Ac ionic liquid before calcination. | ||
Fig. 5 shows the typical XPS survey spectrum of the TiO2 nanosheet sample after calcination. From the Fig. 5, it is found that the as-prepared TiO2 nanosheet sample contains Ti, O and C elements with photoelectron peaks appearing at binding energies of 458 (Ti2p), 530 (O1s) and 285 eV (C1s). The carbon peak can be ascribed to the residual carbon from the sample and adventitious hydrocarbon from the XPS instrument itself.
 | ||
| Fig. 5 XPS survey spectrum of the TiO2 sample synthesized with 87.5 mM [EMIM]Ac ionic liquid after calcination. | ||
In order to understand the influence of [EMIM]Ac concentration on the final products, we perform the synthesis experiments at different concentration of [EMIM]Ac. Fig. 6 shows the typical SEM images of samples obtained at 8.75 mM, and 875 mM [EMIM]Ac before and after calcination. At 8.75 mM [EMIM]Ac, the low and high magnification SEM images clearly show that flowerlike TiO2 particles form (Fig. 6a and b). The size of these flowerlike particles is 501 ± 125 nm. After calcination, it shows that the morphologies of the flowerlike particles do not change obviously (Fig. 6c). The TEM image of the calcined flowerlike particles clearly shows their detailed structure with sheetlike particles as the subunits, demonstrating their porous structure (Fig. 6d). When the concentration of the [EMIM]Ac increases to 875 mM, it shows that porous aggregates form based on the TEM image shown in Fig. 6e. The calcination process still does not influence obviously the morphology of the aggregates as shown in the TEM image in Fig. 6f. Comparing the results of Fig. 2 and 6, it is clear that the concentration of the [EMIM]Ac ionic liquid influences strongly on the morphology of the synthesized particles. The changes of the particle morphologies with different [EMIM]Ac concentration can be attributed to the ionic liquid's capping agent effect for the crystal growth, which is similar to our previous reported nanoparticle-assembled inorganic particles by ionic liquid assisted route.51,52 At low [EMIM]Ac concentration (8.75 mM), the [EMIM]Ac molecules absorb on the surface of initial TiO2 particles. The nanosheets then stack through π–π stack interaction of the neighboring imidazolium rings, which results in flowerlike morphology. When the concentration of [EMIM]Ac increases to 87.5 mM, there is higher amount of the [EMIM]Ac ionic liquid. In this case, the electrostatic repulsion between the cation ions of the [EMIM]Ac weaken the π–π stack interaction of the neighboring imidazolium rings. Therefore, nanosheets form without stacking into flowerlike particles. Further increasing the amount of the [EMIM]Ac to 875 mM, there are largely excess of ionic liquid in the system. In this case, the initial formed TiO2 particles are completely protected by the ionic liquid. Consequently, they aggregate into porous aggregate to reduce the overall surface energy. As a result, the morphology of the synthesized TiO2 particles can be well controlled by the concentration of the [EMIM]Ac ionic liquid.
 | ||
| Fig. 6 SEM (a–c) and TEM (d–f) images of the samples synthesized with different concentration of [EMIM]Ac ionic liquid before (a, b and e) and after calcinations (c, d and f). (a–d): 8.75 mM [EMIM]Ac, (e and f): 875 mM [EMIM]Ac. | ||
The N2 isotherms and pore size distribution of the as-prepared calcined TiO2 particles synthesized at 8.75 mM, 87.5 mM and 875 mM [EMIM]Ac are shown in Fig. 7a–c. The pore size distributions are calculated by using the Barrett–Joyner–Halenda (BJH) method. The average pore size of the flowerlike TiO2 is 3.8, 5.6 and 29.0 nm while the average pore size of the TiO2 nanosheets is 3.8, 7.7 and 16.2 nm. The average pore size of the porous TiO2 aggregates is calculated to be 3.8 and 4.9 nm. The Brunauer–Emmett–Teller (BET) surface area is calculated to be 51.8, 73.5, and 147.8 m2 g−1 for flowerlike particles, nanosheets and porous aggregates, respectively.
 | ||
| Fig. 7 N2 adsorption–desorption isotherms and pore size distribution plots (inset) of the calcined TiO2 samples synthesized with various concentration of [EMIM]Ac ionic liquid: (a) 8.75 mM, (b) 87.5 mM and (c) 875 mM. | ||
To evaluate the photocatalytic activities of the as-prepared calcined flowerlike particles, nanosheets and porous aggregates, the photodegradation of RhB in an aqueous suspension of the particles is tested. For comparison, the commercial Degussa-P25 TiO2 is also tested at the same condition. Degussa-P25 TiO2 is a mixed phase of anatase and rutile which consists of 80% anatase and 20% rutile, and is generally accepted to exhibit exceptionally better photocatalytic efficiency.38 The performance of the studied photocatalysts toward RhB degradation was determined by monitoring the intensity change of the 553 nm peak versus time. Before UV irradiation, the reaction media containing the catalyst and RhB was stirred in the dark for 1 h to ensure adsorption of RhB on the surface of catalyst. All the adsorption capacity of flowerlike particles, nanosheets, porous aggregates, and Degussa-P25 TiO2 is less than 5%, as shown in Fig. 8a. In the blank test without any TiO2 catalyst, RhB was only degraded by 4.6% over 25 min. The plots of ln(C/C0) are shown in Fig. 8b. Based on Fig. 8b, the first-order reaction rate constants (k) can be calculated. As shown in Fig. 8b, the k for flowerlike particles, nanosheets, porous aggregates, and Degussa-P25 TiO2 is 0.138, 0.199, 0.066, and 0.173 min−1, respectively. It is clear that the TiO2 nanosheets show the highest photocatalytic activity than others. Especially, the photocatalytic activity of the as-prepared nanosheets is higher than the commercial Degussa-P25 TiO2. This seems contrary to the results of the surface area for the various catalysts (flowerlike particles: 51.8 m2 g−1, nanosheets: 73.5 m2 g−1, porous aggregates: 147.8 m2 g−1). Based on the surface area results, one might expect that the photocatalytic activity could follow the order: porous aggregates > nanosheets > flowerlike particles. Although the surface area of TiO2 porous aggregate is the highest its photocatalytic activity is not the best. This can be explained as follows. From Fig. 1, it shows that the porous aggregate is not highly crystallized as the nanosheets and flowerlike particles. The worse crystallinity of porous aggregates will weaken its photocatalytic activity since the photocatalytic activity of anatase TiO2 is dependent on its crystallinity.53 Also, the average pore size (3.8, 7.7 and 16.2 nm) of the TiO2 nanosheets is higher than the porous TiO2 aggregates (3.8 and 4.9 nm). This is also beneficial for the higher photocatalytic activity of TiO2 nanosheets in comparison with the porous TiO2 aggregates. Moreover, the porous aggregates cannot disperse well in the reaction medium due to the large size of the aggregates, which also results in the worse photocatalytic activity. The highest photocatalytic activity for nanosheets results from the better crystallinity, relatively higher surface area, larger pore size and highly easier dispersibility. Therefore, the present synthesis route could provide excellent TiO2 catalyst for the promising application towards the degradation of organic molecules. The photocatalytic mechanism of the as-prepared TiO2 can be described as follows.54 During the irradiation of UV light, conduction-band electrons and valence-band holes separate. The holes attack the water or surface hydroxyls and yield highly reactive hydroxyl radicals (˙OH) at the interface. Moreover, photogenerated electrons transferring to the electron donors like O2 absorbed at the TiO2 surface also contribute to the production of large amounts of hydroxyl radicals. The produced hydroxyl radicals can act as effective centers for photocatalytic reactions, resulting in the decomposition of Rhodamine B through direct reaction with the hydroxyl radicals. The hydroxyl radical-based photocatalytic mechanism is further verified by the detection of the formation of hydroxyl radicals based on the photoluminescence technique.55 The terephthalic acid was chosen as a probe molecule since the terephthalic acid can react with ˙OH to form fluorescent 2-hydroxyterephthalic acid (fluorescence emission peak is around 425 nm when excited at 315 nm). Fig. S2 (ESI†) shows the fluorescence emission spectrum (excitation at 315 nm) of terephthalic acid solution with the as-prepared TiO2 nanosheets after UV irradiation for 15 min. A strong fluorescence emission peak at 425 nm appears, demonstrating the formation of the hydroxyl radicals during the UV irradiation process.
 | ||
| Fig. 8 The residual concentration (C/C0) (a) and the mean ln(C/C0) (b) of RhB in aqueous solutions after different UV irradiation time in the presence of various TiO2 catalysts. C0 and C are the equilibrium concentration of RhB before and after UV irradiation, respectively. | ||
Conclusions
In summary, an ionic liquid-assisted synthesis route is presented for the synthesis of anatase TiO2 particles. It demonstrates that flowerlike TiO2 particles, TiO2 nanosheets and porous TiO2 aggregates can be controlled synthesized with the assistance of ionic liquid 1-ethyl-3-methylimidazolium acetate. The photocatalytic activity of the as-prepared TiO2 particles is investigated, which is compared with the commercial Degussa-P25 TiO2. The as-prepared TiO2 nanosheets exhibit the highest photocatalytic activity towards the degradation of RhB molecules, which is attributed to their high crystallinity, high surface area and high dispersibility. Specifically, the photocatalytic activity of TiO2 nanosheets is higher than the commercial Degussa-P25 TiO2, demonstrating their promising application towards the degradation of organic molecules. The present work thus shows that the ionic liquid-assisted synthesis route is very promising for the controllable synthesis of TiO2 with improved photocatalytic activity.Acknowledgements
This work is supported by National Natural Science Foundation of China (Grant No. 21173127), and the Fundamental Research Funds of Shandong University (Grant No. 2015JC003).References
- A. Taubert and Z. Li, Dalton Trans., 2007, 723 RSC.
- Z. Li, Z. Jia, Y. Luan and T. Mu, Curr. Opin. Solid State Mater. Sci., 2008, 12, 1 CrossRef CAS PubMed.
- Z. Ma, J. H. Yu and S. Dai, Adv. Mater., 2010, 22, 261 CrossRef CAS PubMed.
- X. Liu, J. Ma and W. Zheng, Rev. Adv. Mater. Sci., 2011, 27, 43 CAS.
- E. Ahmed, J. Breternitz, M. Friedrich Groh and M. Ruck, CrystEngComm, 2012, 14, 4874 RSC.
- D. Freudenmann, S. Wolf, M. Wolff and C. Feldmann, Angew. Chem., Int. Ed., 2011, 50, 11050 CrossRef CAS PubMed.
- X. F. Zhang, R. S. Kuehnel, H. T. Hu, D. Eder and A. Balducci, Nano Energy, 2015, 12, 207 CrossRef CAS PubMed.
- C. Liu, B. Zhang, J. Zhang, L. Peng, X. Kang, B. Han, T. Wu, X. Sang and X. Ma, Chem. Commun., 2015, 51, 11445 RSC.
- K. Ding, Z. Miao, Z. Liu, Z. Zhang, B. Han, G. An, S. Miao and Y. Xie, J. Am. Chem. Soc., 2007, 129, 6362 CrossRef CAS PubMed.
- A. Guleria, A. K. Singh, M. C. Rath, S. K. Sarkar and S. Adhikari, Dalton Trans., 2014, 43, 11843 RSC.
- H. Kaper, F. Endres, I. Djerdj, M. Antonietti, B. Smarsly, J. Maier and Y. S. Hu, Small, 2007, 3, 1753 CrossRef CAS PubMed.
- Q. Ma, L. Ping, H. Zhang, J. Yang and Q. Wang, J. Mol. Struct., 2015, 1091, 1 CrossRef CAS PubMed.
- E. Delahaye, Z. L. Xie, A. Schaefer, L. Douce, G. Rogez, P. Rabu, C. Günter, J. S. Gutmann and A. Taubert, Dalton Trans., 2011, 40, 9977 RSC.
- Y. Zhu, W. Wang, R. Qi and X. Hu, Angew. Chem., Int. Ed., 2004, 43, 1410 CrossRef CAS PubMed.
- H. Wender, P. Migowski, A. F. Feil, L. F. de Oliveira, M. H. G. Prechtl, R. G. Machado, S. R. Teixeira and J. Dupont, Phys. Chem. Chem. Phys., 2011, 13, 13552 RSC.
- K. Richter, A. Birkner and A. V. Mudring, Angew. Chem., Int. Ed., 2010, 49, 2431 CrossRef CAS PubMed.
- P. Hoang, H. Park and D. Kim, J. Am. Chem. Soc., 2011, 133, 1465 Search PubMed.
- D. Sung and H. Park, J. Colloid Interface Sci., 2011, 357, 46 CrossRef PubMed.
- Y. L. Verma, R. K. Singh, I. K. Oh and S. Chandra, RSC Adv., 2014, 4, 39978 RSC.
- L. Xiao, H. Shen, R. von Hagen, J. Pan, B. Lhoussaine and S. Mathur, Chem. Commun., 2011, 46, 6509 RSC.
- H. Cheng, B. Huang, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem.–Eur. J., 2011, 17, 8039 CrossRef CAS PubMed.
- K. S. Yao, W. W. Lu, X. Y. Li, J. J. Wang and J. L. Yuan, Chem. Commun., 2013, 49, 1398 RSC.
- J. M. Yang, S. P. Gou and I. W. Sun, Chem. Commun., 2010, 46, 2686 RSC.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005 CrossRef CAS PubMed.
- L. Du, Z. Long, H. Wen, W. Ge, Y. Zhou and J. Wang, CrystEngComm, 2014, 16, 9096 RSC.
- H. Wen, Y. Zhou, J. Xie, Z. Long, W. Zhang and J. Wang, RSC Adv., 2014, 4, 49647 RSC.
- Z. Li, A. Geßner, J. Richters, J. Kalden, T. Voss, C. Kubel and A. Taubert, Adv. Mater., 2008, 20, 1279 CrossRef CAS PubMed.
- J. Ma, L. Chang, J. Lian, Z. Huang, X. Duan, X. Liu, P. Peng, T. Kim, Z. Liu and W. Zheng, Chem. Commun., 2010, 46, 50060 Search PubMed.
- Y. J. Zhao, J. L. Zhang, B. X. Han, J. L. Song, J. S. Li and Q. A. Wang, Angew. Chem., Int. Ed., 2011, 50, 636 CrossRef CAS PubMed.
- K. Richter, P. S. Campbell, T. Baecker, A. Schimitzek, D. Yaprak and A. V. Mudring, Phys. Status Solidi B, 2013, 250, 1152 CrossRef CAS PubMed.
- Z. Li, H. Gong, T. Mu and Y. Luan, CrystEngComm, 2014, 16, 4038 RSC.
- Z. Li, J. Li, L. Song, H. Gong and Q. Niu, J. Mater. Chem. A, 2013, 1, 15377 CAS.
- Z. Lu, M. Ye, N. Li, W. Zhong and Y. Yin, Angew. Chem., Int. Ed., 2010, 49, 1862 CrossRef CAS PubMed.
- H. G. Moon, Y. S. Shim, H. W. Jang, J. S. Kim, K. J. Choi, C. Y. Kang, J. W. Choi, H. H. Park and S. J. Yoon, Sens. Actuators, B, 2010, 149, 116 CrossRef CAS PubMed.
- S. Ding, J. S. Chen, Z. Wang, Y. L. Cheah, S. Madhavi, X. Hu and X. W. Lou, J. Mater. Chem., 2011, 21, 1677 RSC.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissortel, J. Salbeck, H. Spreitzer and M. Gratzel, Nature, 1998, 395, 583 CrossRef CAS.
- M. Ye, Q. Zhang, Y. Hu, J. Ge, Z. Lu, L. He, Z. Chen and Y. Yin, Chem.–Eur. J., 2010, 16, 6243 CrossRef CAS PubMed.
- H. Y. Liu, J. B. Joo, M. Dahl, L. S. Fu, Z. Z. Zeng and Y. D. Yin, Energy Environ. Sci., 2015, 8, 286 CAS.
- Y. Zhou and M. Antonietti, J. Am. Chem. Soc., 2003, 125, 14960 CrossRef CAS PubMed.
- T. Nakashima and N. Kimizuka, J. Am. Chem. Soc., 2003, 125, 6386 CrossRef CAS PubMed.
- W. J. Zheng, X. D. Liu, Z. Y. Yan and L. J. Zhu, ACS Nano, 2009, 3, 115 CrossRef CAS PubMed.
- H. Wang, X. Tan and T. Yu, Appl. Surf. Sci., 2014, 321, 537 CrossRef PubMed.
- J. Yu, Q. Li, S. Liu and M. Jaroniec, Chem.–Eur. J., 2013, 19, 2433 CrossRef CAS PubMed.
- F. Mirhoseini and A. Salabat, RSC Adv., 2015, 5, 12536 RSC.
- R. Ramanathan and V. Bansal, RSC Adv., 2015, 5, 1424 RSC.
- A. Wittmar, H. Thierfeld, S. Köchera and M. Ulbricht, RSC Adv., 2015, 5, 35866 RSC.
- Q. Xu, J. Yu, J. Zhang, J. Zhang and G. Liu, Chem. Commun., 2015, 51, 7950 RSC.
- S. M. Lee, S. N. Cho and J. W. Cheon, Adv. Mater., 2003, 15, 441 CrossRef CAS PubMed.
- Z. Li, A. Friedrich and A. Taubert, J. Mater. Chem., 2008, 18, 1008 RSC.
- J. Dupont, J. Braz. Chem. Soc., 2004, 15, 341 CrossRef CAS.
- Z. Li, R. Li, T. Mu and Y. Luan, Chem.–Eur. J., 2013, 19, 6005 CrossRef CAS PubMed.
- R. Li, J. Du, Y. Luan, H. Zou, G. Zhuang and Z. Li, CrystEngComm, 2012, 14, 3404 RSC.
- T. J. Zhu, J. Li and Q. S. Wu, ACS Appl. Mater. Interfaces, 2011, 3, 3448 CAS.
- F. Xiao, J. Mater. Chem., 2012, 22, 7819 RSC.
- J. Yu, W. Wang, B. Cheng and B. L. Su, J. Phys. Chem. C, 2009, 113, 6743 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17213f |
| This journal is © The Royal Society of Chemistry 2015 |