
ACdCO3F (A = K and Rb): new noncentrosymmetric materials with remarkably strong second-harmonic generation (SHG) responses enhanced via π-interaction†
Abstract
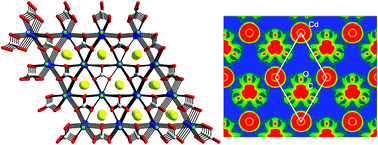
Two new noncentrosymmetric (NCS) materials, namely, ACdCO3F (A = K and Rb) containing both a d10 cation (Cd2+) and a π-conjugated parallel carbonate anion (CO32−), were synthesized through conventional solid state reactions. ACdCO3F exhibits a 3-dimensional structure that is composed of the stacked layers of [Cd(CO3)]∞. Each [Cd(CO3)]∞ layer is connected by infinite Cd–F–Cd chains and the [CO3] triangles are oriented in the same direction with a coplanar alignment. KCdCO3F and RbCdCO3F reveal remarkably strong second-harmonic generation (SHG) responses of approximately 9.0 and 7.2 times that of potassium dihydrogen phosphate (KDP), respectively, and both materials are phase-matchable. ACdCO3F exhibit wide transparent regions ranging from far UV to mid IR. Theoretical calculations confirm that the large SHG efficiencies indeed originate from enhancement via interatomic interactions between the s and p states of Cd2+ and the π-conjugated groups of the [CO3]2− unit within the [Cd(CO3)]∞ layers.
 Please wait while we load your content...
Please wait while we load your content...

