
Anatase TiO2 nanoparticles with controllable crystallinity as a substrate for SERS: improved charge-transfer contribution
Abstract
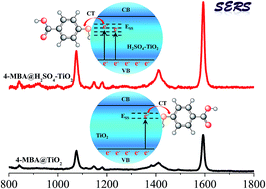
In this paper, TiO2 nanoparticles (NPs) in the anatase phase were synthesized by a very simple sol-hydrothermal method with the assistance of H2SO4. And importantly, the crystallinity and the surface defects (surface state) of anatase TiO2 NPs can be regulated and controlled by this means for improving their SERS performances. The 4-MBA molecule adsorbed on 0.14 mL-H2SO4–TiO2 NPs exhibits the highest SERS enhancement, as compared with the native enhancement on unmodified TiO2 NPs. The introduction of H2SO4 not only enriches the surface defects of anatase TiO2 and the adsorption site, but also improves the separation efficiency of photo-generated carriers, which all can promote the TiO2-to-molecule charge transfer and are mainly responsible for the improved SERS performances.
- This article is part of the themed collection: Surface enhanced Raman Spectroscopy: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

