
Regulating the size and molecular weight of polymeric particles by 1,1-diphenylethene controlled soap-free emulsion polymerization†
Abstract
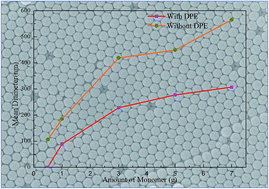
We herein report a facile soap-free emulsion polymerization (SFEP) method to prepare living particles with size and molecular weight that could be easily regulated. In this method, commercially available, odorless, and colorless 1,1-diphenylethene with no known toxicity was introduced to decrease the size and molecular weight of the particles. Using styrene as a model monomer, a series of monodisperse PS particles with a mean diameter ranging from 89 nm to 307 nm and a molecular weight ranging from 613 g mol−1 to 11 760 g mol−1 were produced via adjusting the amount of the monomer and DPE added into the SFEP system.
 Please wait while we load your content...
Please wait while we load your content...

