
Pressure evolution of the potential barriers for transformations of layered BN to dense structures
Abstract
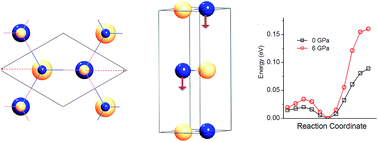
Layered BN is a graphite analogue with exceptionally strong covalent bonding, while dense phases, particularly cubic and wurtzite BN are important hard materials for machine tools and other applications. BN is invariably formed in the layered form and must be converted to the dense phase under pressure. We report the pressure dependent structure of BN and energy barriers for the transformations as a function of initial structure based on first-principles methods including dispersion interactions, which we find to be important. The cohesive energies of the layered structures are similar to that of wurtzite BN. The energy barriers for transformation from rhombohedral layered BN to dense cubic BN and that from hexagonal layered BN to dense wurtzite BN are found to be similar at low pressure. Increasing pressure results in a rapid decrease of these energy barriers. The phase transition from ordered layered structures to the dense phase should be completed at approximately 25 GPa. We find a large energy barrier for layer glides under pressure, which provides an explanation for the difficulty in ambient temperature, high pressure formation of cubic dense BN from layered starting materials that contains stacking faults.
 Please wait while we load your content...
Please wait while we load your content...

